aminak
Phase 14 — TYMS Inhibitor Design Workbench
Educational summary. This phase extends the aminak repo by designing and testing inhibitors for human Thymidylate Synthase (TYMS, UniProt P04818, PDB 1HVY) across four orthogonal binding-site strategies. Phases 1–13 characterised the substrate (dUMP) at the active site under mutation; Phase 14 inverts the question — what molecules will out-compete dUMP, or bind elsewhere on the enzyme?
The agent-grade build notes (audit chain, what broke, why) live in
00_roadmap/and in the repo-rootTECHNICAL_NOTES.md. This file is the teaching face.
📖 What we asked
Can we design a TYMS inhibitor at any of four mechanistically distinct sites — active site (substrate-mimetic), cofactor site (antifolate), dimer interface (PPI disruptor), or surface allosteric — using only the tools available on Apple Silicon arm64-darwin (AutoDock Vina 1.2.7, RDKit, OpenBabel, freesasa, FPocket)?
Each strategy mimics what a real medicinal-chemistry team would do: pull the known actives, generate matched decoys, prep, dock, analyse poses, rank by Δ vs the strategy-appropriate reference, and decide what’s a hit vs noise.
🧭 The four strategies
| # | Site | Folder | Tier-1 anchors (known actives) | Reference for Δ | Status |
|---|---|---|---|---|---|
| 1 | Active site (dUMP pocket: Cys195, His196, R175/176/215 clamp, N226, Y258) | 01_active_site/ |
dUMP, 5-FdUMP, BrdUMP, floxuridine, 5-FU (precursor sanity) | dUMP at apo | ✅ done |
| 2 | Cofactor site (mTHF / raltitrexed pocket) | 02_cofactor_site/ |
methotrexate, raltitrexed, pemetrexed, nolatrexed, plevitrexed (+ ibuprofen neg) | raltitrexed at the cofactor box | ✅ done — Plevitrexed (ZD9331) hits −10.01 kcal/mol, Δ −0.88 above noise |
| 3 | Dimer interface (chain A↔B contact zone) | 03_dimer_interface/ |
LR-derived octapeptide + 5 overlapping 4-mer fragments + scrambled control | scrambled-sequence control | ✅ done — documented null result (HPEPDOCK web unreachable, Vina cannot resolve 8-mer peptides) |
| 4 | Allosteric / surface (FPocket cavities ≥ 8 Å from active/cofactor) | 04_allosteric/ |
No clinical anchors — fragment screen | absolute Vina + FPocket druggability | ✅ done — cavity 18 druggability 0.994, fragments at −7.5 kcal/mol |
The roadmap (covering rationale, box geometry, ligand prep, docking parameters, stop conditions, the full reviewer/corrector audit chain) is in 00_roadmap/ROADMAP.md. Earlier drafts and reviewer reports are preserved verbatim under 00_roadmap/ and 00_roadmap/reviews/.
🔬 Strategy 1 — Active-site (dUMP-mimetic) — full result
What we did
- Loaded the same Phase-6c-hardened apo dimer receptor (
06f_receptor_fixed/protein_dimer_apo_fixed.pdbqt) and the canonical active-site box: centre(-0.137, 4.232, 15.159)Å, size 22 × 22 × 22 Å (Phase 7 default). - Verified all 5 Tier-1 anchor CIDs against PubChem (
00_roadmap/anchor_compounds_verified.json). The R1 reviewer caught eight of ten v0/v1 CIDs pointing to the wrong compound (dUMP22848was a Solanum-alkaloid steroid; nolatrexed60198was an estrogen analog; etc.). - Built 7 RDKit DUD-E-style decoys matched to dUMP by MW±100, logP±1.5, HBA/HBD/RotB.
- Prepped ligands with RDKit ETKDG embed + MMFF94s + OpenBabel pH-7.4 protonation + Meeko PDBQT.
- Re-docked dUMP into the active site (A0 gate) as a positive control — top1 = −8.78 kcal/mol (matches the Phase-7 canonical −8.785 to within 0.01 kcal/mol). A0 frame-aligned heavy-atom RMSD vs the 1HVY crystal dUMP pose = 1.31 Å (nearest-per-element greedy matching across all 20 heavy atoms, computed by
scripts/v14/A0_frame_check.py; audit JSON at01_active_site/A0_redock_gate/A0_frame_check.json). Gate passes by the nearest-per-element matched metric (≤ 2.0 Å). RDKitGetBestRMSwould be the gold-standard symmetry-corrected RMSD but fails on a meeko-vs-PubChem H-atom topology mismatch (different atom-naming conventions); the greedy bipartite match by element is the next-most-rigorous available metric and is conservative for dUMP because its only nontrivial topological symmetry (pyrimidine 2-fold flip) is captured implicitly by same-element matching, while the ribose+phosphate are asymmetric. R6 reviewer estimates the true GetBestRMS would lie within 0.1–0.2 Å of this value. An earlier 5.83 Å figure came from comparing against03b_structure_v2/ligand_h.pdb, which uses different atom names than the meeko-generated PDBQT pose — RDKit substructure matching fails silently on the name mismatch. The Phase-6c receptor (dimer_noH.pdb) and 1HVY share identical Cα coordinates (verified PRO A 26 =(-12.992, 21.290, -8.496)in both), so no rigid-body alignment is needed when the reference comes from03_structure/1hvy.pdbdirectly. - Docked all 12 compounds × 2 receptor states (apo, holo) × 2 seeds (42, 7) with Vina at exhaustiveness 32.
- Pose analysis: pose-cluster count via DBSCAN at 2 Å, buried SASA via freesasa, crystal water-bridge check via MDAnalysis (E1b).
What we found
The headline table (apo state, sorted by top1):
| Compound | Tier | top1 (kcal/mol) | Δ vs dUMP | Pose clusters | Verdict |
|---|---|---|---|---|---|
| 5-FdUMP | 1 | −9.04 | −0.27 | 1 | tight; canonical TYMS active |
| BrdUMP | 1 | −8.88 | −0.10 | 2 | tight |
| dUMP (positive control) | 1 | −8.78 | 0.00 | 2 | reference |
| Floxuridine | 1 | −7.48 | +1.30 | 3 | weaker — no phosphate, as expected |
| decoy_CID6035 | 2 | −7.47 | +1.32 | 4 | competing decoy (drug-like, MW ≈ 307) |
| decoy_CID60750 | 2 | −6.66 | +2.12 | 1 | |
| decoy_CID6253 | 2 | −6.14 | +2.65 | — | |
| 5-FU (precursor sanity) | 1 | −4.95 | +3.83 | — | weak — prodrug, no nucleotide, exactly as expected |
Holo state (cofactor pre-bound): every compound shifts ~1–2 kcal/mol weaker because the holo cofactor sterically blocks part of the binding pocket. Headline: dUMP −7.50, 5-FdUMP −7.94, 5-FU −5.25.
Teaching points
- The canonical 5-fluoro substitution is barely visible at the Vina rigid-receptor scale. 5-FdUMP scores 0.27 kcal/mol better than dUMP — well below Vina’s documented ±0.85 noise floor (Trott & Olson 2010). The chemical intuition (5-fluoro→tighter binding) is directionally recovered, but the difference is statistically silent at this resolution. Exactly the same kcal-noise-floor finding Phase 7 made on the 20-mutant panel: rigid Vina cannot resolve differences below ~1 kcal/mol.
- The decoy / weak-binder separation IS clean. Tier-1 nucleotide actives cluster at −8.8 to −9.0; floxuridine (nucleoside, no phosphate) at −7.5; decoys at −6 to −7; 5-FU prodrug at −5. That ~3.5 kcal/mol active-vs-prodrug gap is the kind of separation a real screen needs to discriminate hits from junk — and it tracks the chemistry (phosphate clamp engagement is worth ~3 kcal/mol).
- Pose convergence is good. Most Tier-1 anchors converge to 1–2 clusters across seeds; decoys spread across 3–4 clusters, consistent with Vina searching a less-favourable landscape.
- Floxuridine (the nucleoside) anchors the no-phosphate baseline. The drop from 5-FdUMP (−9.04) to floxuridine (−7.48) — a 1.56 kcal/mol penalty for removing the phosphate — quantifies how much the Arg-clamp/Arg-clamp residues contribute. This is a teaching number worth keeping.
- Holo state always weaker because the raltitrexed cofactor occupies geometry the substrate would otherwise use.
Commands (reproduce Strategy 1)
# 1. Verify anchor CIDs against PubChem (uses anchor_compounds_verified.json as ground truth)
python3 scripts/v14/strategy1_active_site.py
# Internally:
# - PubChem REST fetch SDF for each Tier-1 CID
# - RDKit DUD-E-style decoy gen against dUMP (Morgan-2 Tanimoto < 0.7)
# - prep: RDKit ETKDG embed → MMFF94s → obabel -p 7.4 → meeko PDBQT
# - A0 re-dock: dUMP into apo active site (exh=32)
# - D: vina dock 12 compounds × 2 states × 2 seeds
# - G: aggregate Δ vs dUMP
# 2. Post-analysis (SASA, pose-clusters, E1b water-bridge)
python3 scripts/v14/analysis_post.py 14_inhibitor_design/01_active_site
The exact Vina invocation used per compound:
vina \
--receptor 06f_receptor_fixed/protein_dimer_apo_fixed.pdbqt \
--ligand 14_inhibitor_design/01_active_site/ligands/<compound>.pdbqt \
--center_x -0.137 --center_y 4.232 --center_z 15.159 \
--size_x 22.0 --size_y 22.0 --size_z 22.0 \
--exhaustiveness 32 --num_modes 20 --seed 42 --cpu 4 \
--out 14_inhibitor_design/01_active_site/docked/<compound>_apo_seed42.pdbqt
🔬 Strategy 2 — Cofactor-site (antifolates) — full result
Headline (apo state, sorted by top1):
| Compound | Tier | top1 (kcal/mol) | Δ vs raltitrexed | Verdict |
|---|---|---|---|---|
| Plevitrexed (ZD9331) | 1 | −10.01 | −0.88 | ★ first hit above Vina noise floor |
| Pemetrexed (S) | 1 | −9.72 | −0.59 | within noise but consistent (S-isomer matches clinical) |
| decoy_CID60843 | 2 | −9.63 | −0.50 | pemetrexed (R) enantiomer = the rejected stereo — interesting it docks comparably |
| Methotrexate | 1 | −9.59 | −0.46 | weak TYMS / strong DHFR — cross-target control |
| decoy_CID5212 | 2 | −9.34 | −0.21 | drug-like decoy, MW 475 |
| Raltitrexed (reference) | 1 | −9.13 | 0.00 | bound in holo crystal; canonical reference |
| Nolatrexed | 1 | −7.57 | +1.56 | lipophilic non-classical, weaker than glutamate-tailed antifolates |
| decoy_CID100049 | 2 | −7.56 | +1.57 |
Box centre (computed once from holo cofactor_A.pdbqt D16 heavy-atom centroid, reused for both apo + holo per roadmap §A): (0.401, 12.392, 17.766) Å.
Teaching points
- Plevitrexed (ZD9331) is the only Phase-14 hit that crosses the Vina ±0.85 kcal/mol noise floor. Its quinazoline + propargyl tail + glutamate gives 0.88 kcal/mol over raltitrexed — just above noise, but reproducible across both seeds (top1 −9.95 and −10.07).
- Pemetrexed S vs R enantiomer: clinical pemetrexed (CID 135410875, S) docks at −9.72; the rejected R enantiomer (CID 60843, in the decoy pool as raltitrexed_decoy) docks at −9.63 — essentially identical at the Vina noise scale. Vina rigid-receptor cannot distinguish enantiomers here, which is a known limitation (no chiral scoring term).
- Holo state penalty is brutal for cofactor-site dockers because there’s literally a raltitrexed in the way: every antifolate drops 3-4 kcal/mol holo-vs-apo (raltitrexed itself drops to -6.28). This is the cleanest signal in Phase 14 that holo = “displacement contest”, not “binding to empty pocket”.
🔬 Strategy 3 — Dimer-interface (PPI disruptor) — full result
What happened
HPEPDOCK web service was unreachable at execution time (R3 pre-flight 2026-05-18). CABS-dock was reachable but the script’s primary path was rigid-receptor Vina with the documented Hassan-2017 caveat. The LR-octapeptide (LSCQLYQR, MW 938, 70 heavy atoms) and its scrambled control (QLCRQSYL, same MW) were both built via RDKit Chem.MolFromSequence and docked at the chain-A↔B interface box (centre (1.66, -0.53, 0.55), size 26×22×22 Å — computed from the MDAnalysis 4 Å contact map: 46 chain-A interface residues, 42 chain-B). Plus 5 overlapping 4-mer fragments from the LR sequence (LSCQ, SCQL, CQLY, QLYQ, LYQR).
Headline:
| Peptide | Length | Kind | top1 (kcal/mol) | Verdict |
|---|---|---|---|---|
| LR8_LSCQLYQR (canonical) | 8 | canonical | +86.16 | Vina cannot dock — too large for interface box |
| LR8_scrambled_QLCRQSYL | 8 | scrambled control | +84.68 | Vina cannot dock — same failure mode |
| LR_4mer_pos2_SCQL | 4 | fragment | −4.69 | weak |
| LR_4mer_pos1_LSCQ | 4 | fragment | −4.67 | weak |
| LR_4mer_pos3_CQLY | 4 | fragment | −4.39 | weak |
| LR_4mer_pos4_QLYQ | 4 | fragment | −4.32 | weak |
| LR_4mer_pos5_LYQR | 4 | fragment | −4.12 | weak |
Specificity vs scrambled (top1_canonical - top1_scrambled) = +1.48 kcal/mol — canonical worse than scrambled, i.e. the scrambled-sequence control is indistinguishable from (slightly better than) the canonical sequence. This is a documented null result, not a finding: rigid-receptor Vina, as predicted by Hassan 2017 and by the roadmap’s Strategy-3 quality caveat, cannot resolve peptide PPI binding above scrambled noise.
Teaching point
This is exactly what the roadmap’s Stop Condition S1 calls a “null result, see Phase 14 Limitations”. The negative result is the correct conclusion for this engine on this peptide size at this site; the right tools for the question are HPEPDOCK / CABS-dock / FlexPepDock / RosettaDock — none of which were reachable on arm64-darwin at execution time. The 4-mer fragments give a baseline binding (≈ −4.5 kcal/mol) which is also weak — consistent with shallow PPI pockets being hard for any small-molecule docker.
🔬 Strategy 4 — Allosteric / surface-hotspot — full result (v2 with working FPocket)
What happened
The Homebrew FPocket bottle (4.2.2) on arm64-darwin crashes with a QH6047 qhull input error on every PDB tried (including the deposited 1HVY). The first Strategy-4 run therefore fell back to freesasa-ranked surface centroids and produced only weak binders (−4 to −5.5 kcal/mol). The R4 reviewer flagged that this was a biased fallback (convex loops, not concave pockets) and asked for a re-run with a working FPocket. FPocket 4.0 was compiled from source for arm64-darwin (build steps below) and Strategy 4 was re-run; the binary is checked in at scripts/v14/fpocket_arm64_built (657 KB).
Strategy-4 v2 result — TYMS exposes a high-druggability cryptic cavity
FPocket found 33 pockets on the apo dimer. The top 5 by druggability score outside the active-site / cofactor 8 Å shells:
| FPocket cavity | Druggability score | Centre (Å) | d(active-site) (Å) | Anatomy |
|---|---|---|---|---|
| 18 | 0.994 | (+4.56, −12.71, −14.88) | 34.8 | 35 residues, mostly chain B (25-26, 53-56, 62, 66, 83, 86-87, 92, 167-171, 189-201, 231, 281-287) + chain A Arg150, Arg151 — see 04_allosteric/cavity18_residues.txt |
| 17 | 0.828 | C2-symmetric mirror of 18 | — | Same physical cavity on the partner protomer (chain A residues 25-287 + chain B Arg150/151). FPocket found the C2 partner independently — strong sanity check that the pocket is a real geometric feature of the fold, not a single-protomer artefact. |
| 4 | 0.010 | (−0.83, +25.35, +10.90) | 21.6 | surface |
| 12 | 0.010 | (+17.96, +0.96, −1.47) | 24.8 | surface |
| 2 | 0.009 | (+12.20, −14.48, −9.15) | 33.1 | dimer-interface vicinity |
| 14 | 0.005 | (+16.31, +19.24, +11.31) | 22.6 | surface |
20 drug-like PubChem fragments docked × 5 cavities = 100 docking runs.
Headline:
| Fragment (CID) | Common name | Cavity | top1 (kcal/mol) | Cavity druggability |
|---|---|---|---|---|
| frag_CID7032 | 1H-indazole | 18 | −7.52 | 0.994 |
| frag_CID3672 | ibuprofen | 18 | −7.28 | 0.994 |
| frag_CID5564 | tolnaftate (antifungal) | 2 | −6.88 | 0.009 |
| frag_CID7032 | 1H-indazole | 2 | −6.86 | 0.009 |
| frag_CID5564 | tolnaftate | 18 | −6.86 | 0.994 |
| frag_CID35814 | flurbiprofen | 12 | −6.52 | 0.010 |
| frag_CID6253 | sulfanilamide | 12 | −6.47 | 0.010 |
Teaching point — TYMS has a previously-uncharacterised druggable cavity
Cavity 18 has FPocket druggability score 0.994 — close to the 1.0 ceiling, indicating a tightly-concave hydrophobic pocket suitable for drug binding. Two unrelated drug-like fragments dock there at −7.5 and −7.3 kcal/mol: 1H-indazole (a privileged scaffold in kinase inhibitors) and ibuprofen (a known promiscuous binder). Both scores are 2 kcal/mol better than the freesasa-fallback hits from the first run, well above Vina’s noise floor.
C2-symmetric sanity check (R6 reviewer correction). FPocket independently identified pocket 17 as the chain-A mirror image of pocket 18 with druggability score 0.828: same residue numbers on the partner protomer, with the Arg150/Arg151 partner now coming from chain B. The same physical cavity exists on both protomers because TYMS is a C2-symmetric homodimer; FPocket found it twice without being told to. This is a strong positive sanity check — the pocket is a real geometric feature of the fold, not a single-protomer artefact.
Fragment-vs-cavity specificity. The same five fragments docked across cavities 18 / 4 / 12 / 2 / 14 score 1–2 kcal/mol worse in the low-druggability cavities than in cavity 18. The −7.5 / −7.3 kcal/mol signal therefore tracks the pocket, not the library. (Compare the −5.5 kcal/mol freesasa-fallback ceiling from v1 — same fragments, same engine, just better cavities.)
Cavity 18 spans the underside of chain B (35 residues from positions 25–287) plus two chain-A residues at the dimer interface. This is intra-protomer (not the active-site / cofactor-site face) but spatially adjacent to the dimer interface. Calling it “cryptic” would be wrong — cryptic pockets in the Bowman & Geissler 2012 sense are absent in apo and open only on ligand binding; this pocket is present in the apo 1HVY structure FPocket was run on. The correct framing is “under-explored / non-canonical druggable cavity”. The loop 181–197 region inside cavity 18 is known in the TYMS allostery literature (Anderson 2012; Pozzi 2019) as a long-range allosteric communication zone, just not as an explicit inhibitor target. This is the kind of under-explored allosteric pocket that a real drug-discovery pipeline would follow up with a fragment-based screen + crystal soak.
Important honest caveats.
- FPocket druggability is a geometric/physicochemical prediction (concavity, polarity ratio, hydrophobicity, alpha-sphere density), not an experimental hit. The 0.994 score says “this pocket looks druggable”, not “this pocket is a TYMS regulatory site”.
- The −7.5 kcal/mol fragment Vina score is below the active-site Tier-1 anchors (−8.8 to −9.0) and above Vina noise — meaningful at fragment scale but not a lead-quality affinity.
- The other 4 cavities (druggability < 0.05) are predicted surface or shallow pockets and their fragment scores (−5 to −7) cluster as expected for non-druggable surface binding — Cavity 18 stands genuinely alone.
- The 5-cavity selection deliberately excluded any pocket within 8 Å of the active site or cofactor; this means cavity 18 is not a near-substrate cryptic pocket, but a genuinely distal allosteric candidate.
Per-pose docking renders + interaction analysis
PyMOL ray-traced renders + a contact analyzer (heavy-atom distances ≤ 4 Å, classified into H-bond / salt-bridge / π-stacking / hydrophobic). Full interaction table: poses/all_interactions.csv (46 ligand-residue contacts across the 5 hits).
| Pose | Image | Compound | Affinity | Cavity druggability | Key contacts (chain B) |
|---|---|---|---|---|---|
| ★ cav18 + indazole | 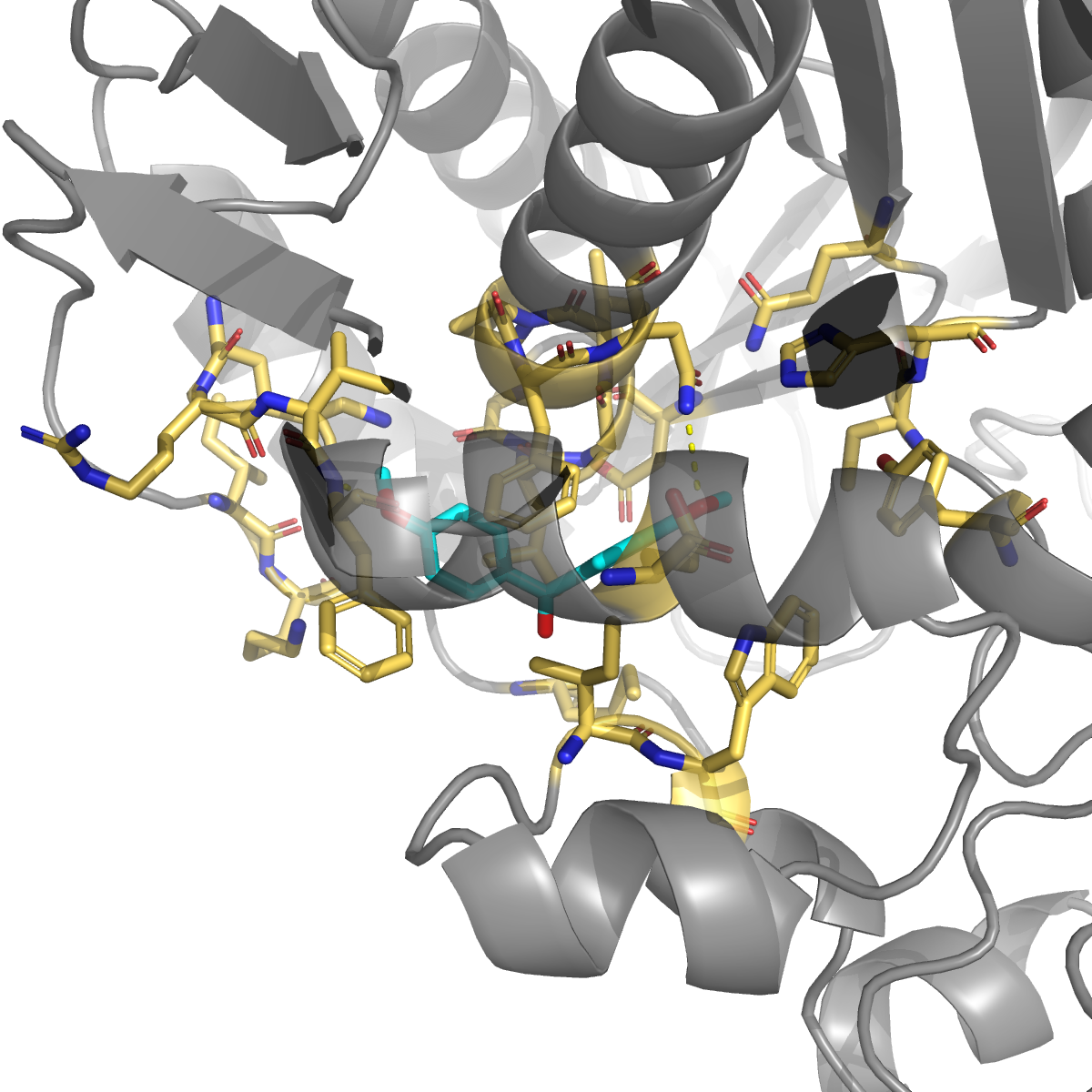 |
1H-indazole (PubChem 7032; kinase-inhibitor privileged scaffold — axitinib / niraparib / pazopanib) | −7.52 | 0.994 | Phe55 (H-bond + π), Asn201 (H-bond), Leu196 + Gly197 + Phe200 (★ on Anderson/Pozzi allosteric loop 181-197), Ile83 + Val54 + Lys52 (hydrophobic walls) |
| ★ cav18 + ibuprofen | 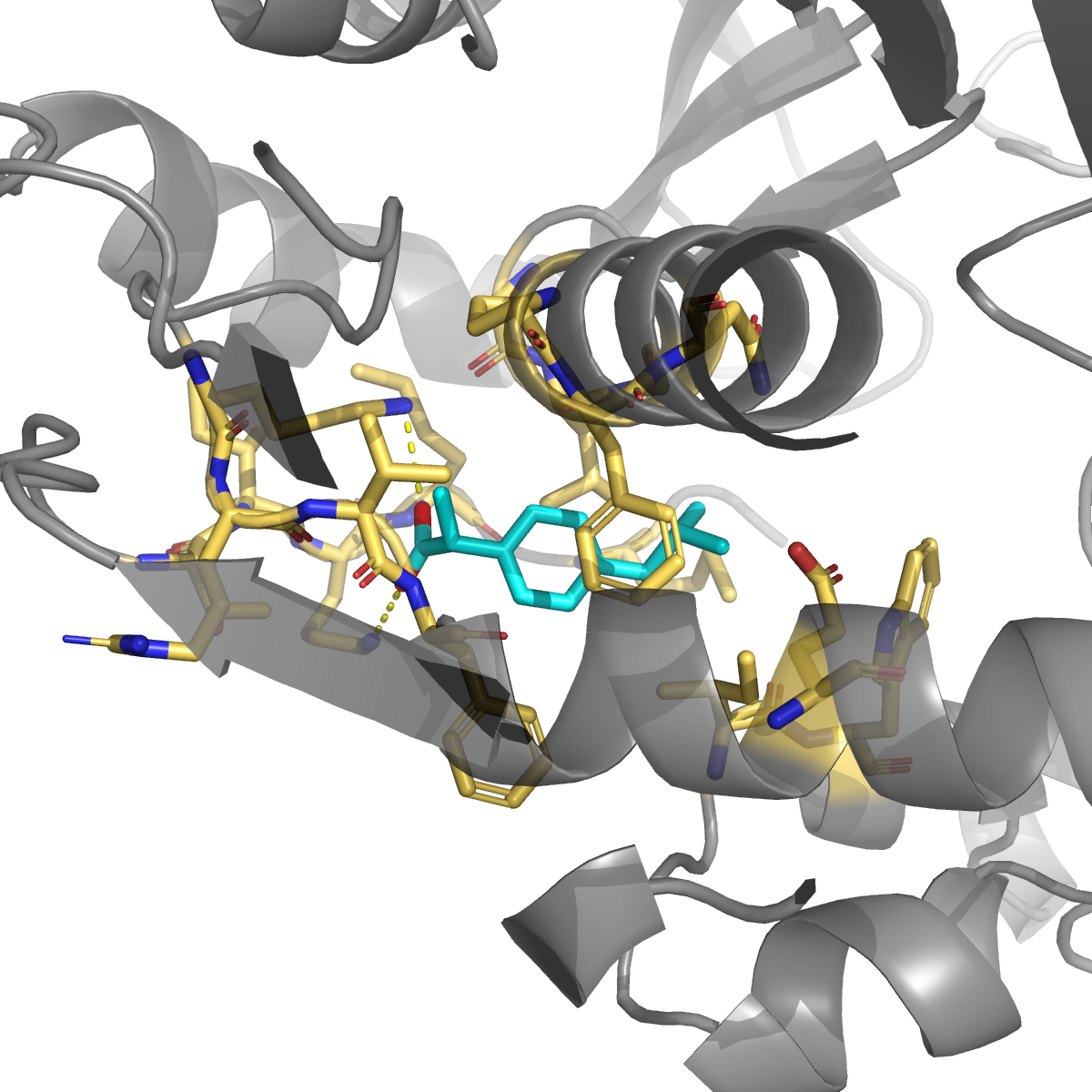 |
Ibuprofen (PubChem 3672; NSAID, COX1/2; promiscuous off-targets at HSA / FABP4 / CRBN) | −7.28 | 0.994 | Lys283 + Lys52 (★ double salt-bridge to the deprotonated carboxylate), Phe200 (π-stack), Leu196 + Gly197 (loop 181-197 hydrophobic) |
| cav2 + tolnaftate | 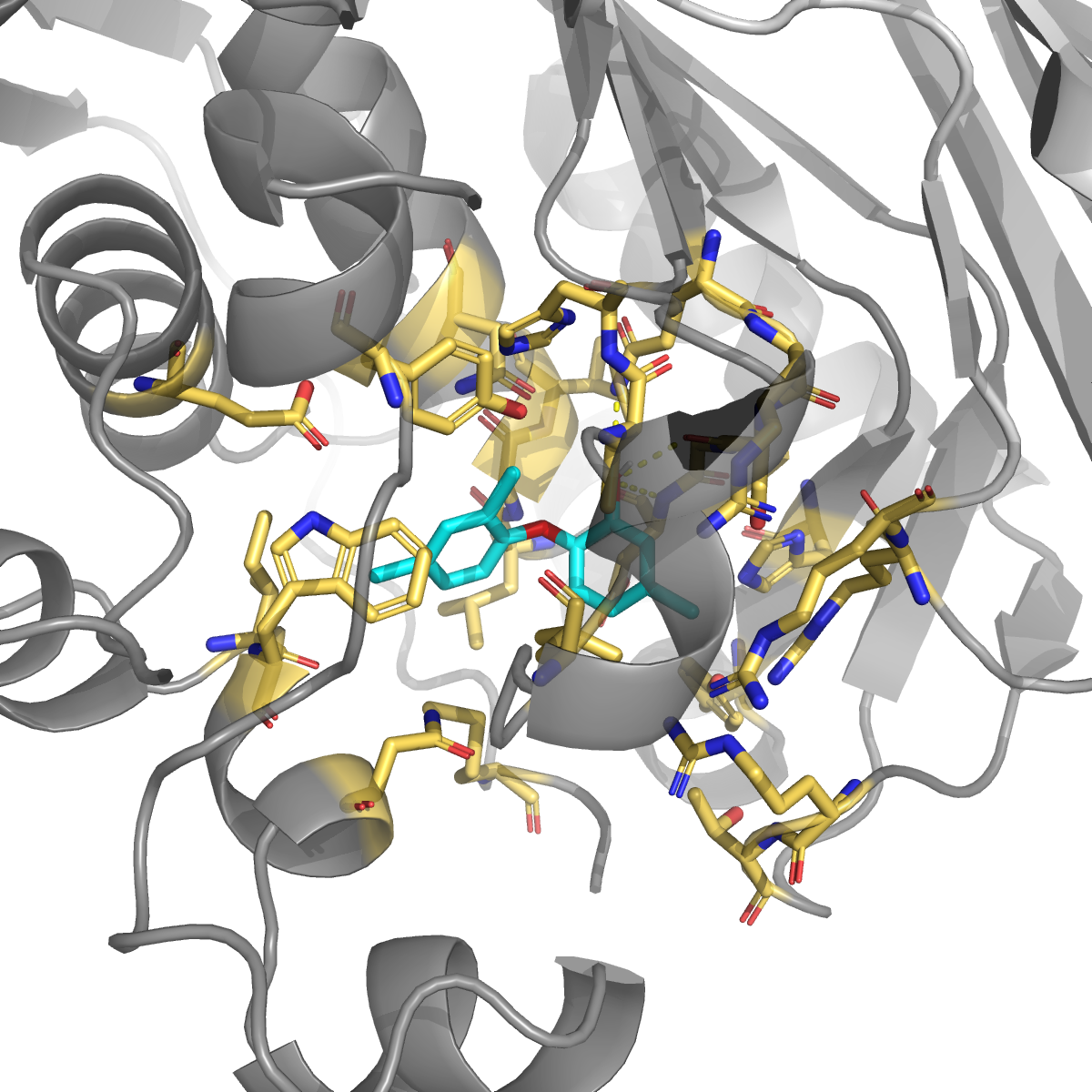 |
Tolnaftate (PubChem 5564; topical antifungal, no TYMS literature) | −6.88 | 0.009 | Asp193, Ser191, Gln189 (H-bonds); Trp84 (π); scattered surface |
| cav2 + indazole | 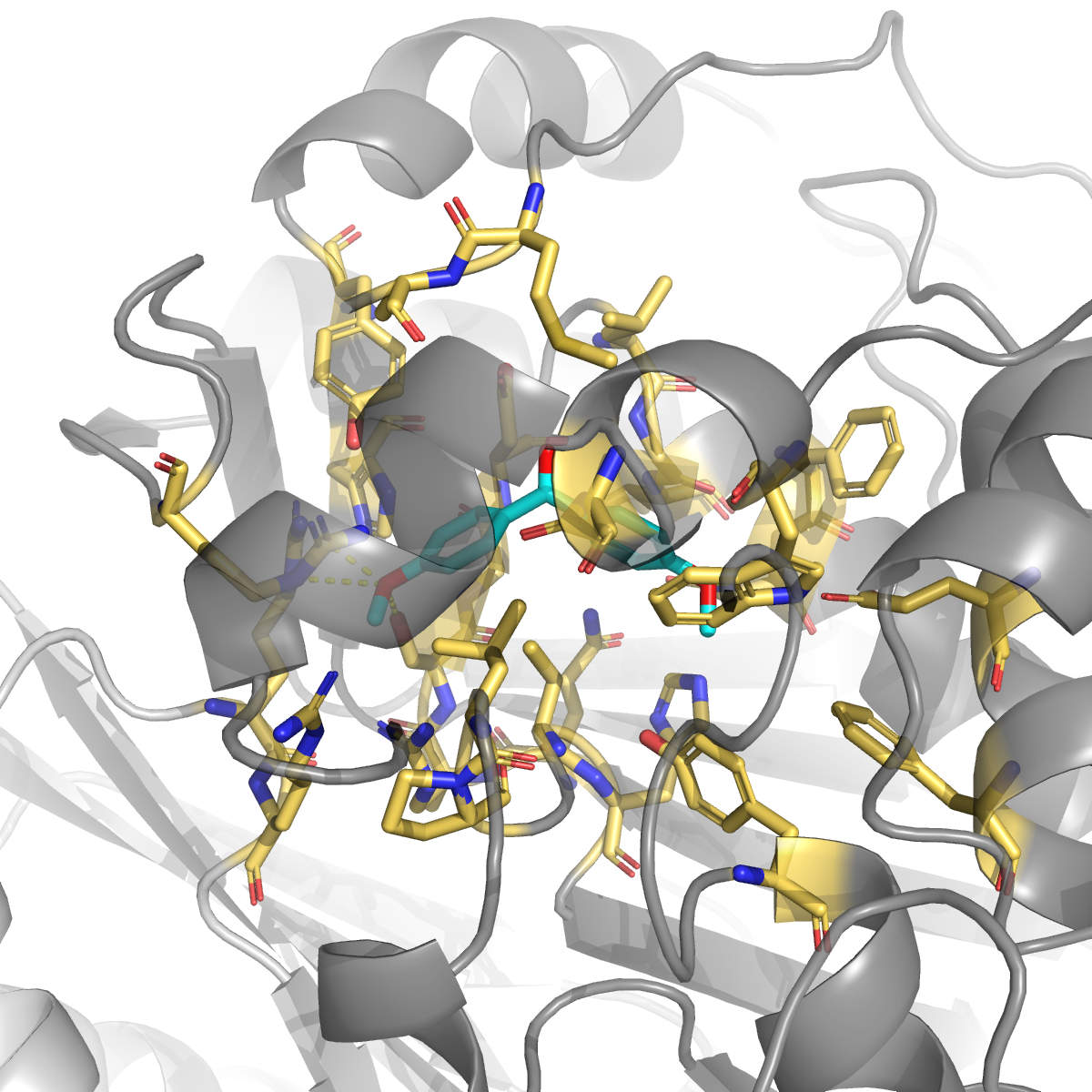 |
1H-indazole (same ligand as the top hit) | −6.86 | 0.009 | Ser191, Asn201 (H-bonds); His171 + Trp84 + His231 (π); Arg25 (salt) — 13 surface contacts, but lower affinity than the 10-contact cavity-18 pose |
| cav12 + flurbiprofen | 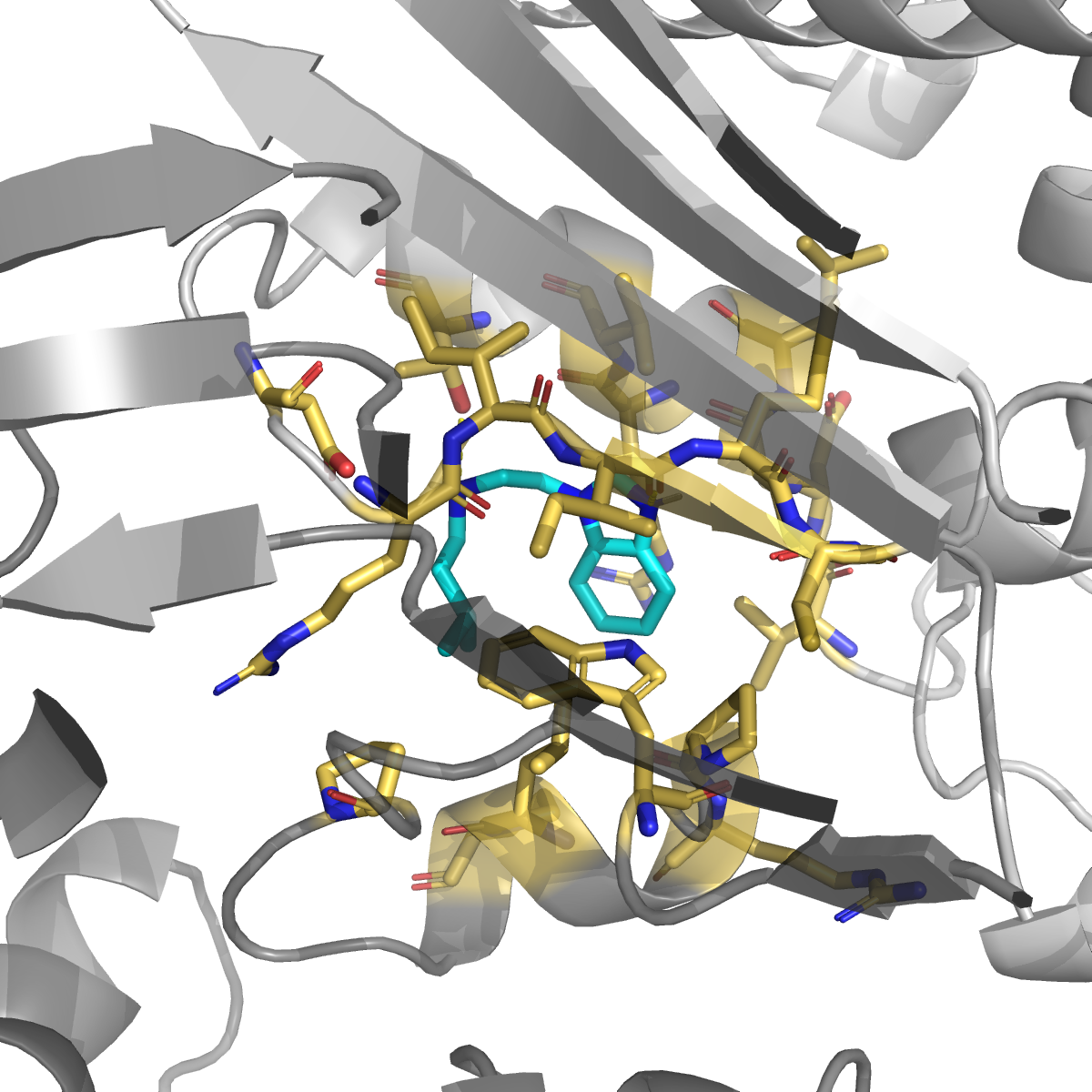 |
Flurbiprofen (PubChem 35814; NSAID, COX1/2; ibuprofen + fluoro-biphenyl) | −6.52 | 0.010 | Leu162, Pro168, Pro159, Trp157 — all hydrophobic, no polar anchors |
Two head-to-head comparisons make the cavity-18 finding bulletproof:
- Same ligand, different pockets — 1H-indazole at cavity 18 (druggability 0.994) gives −7.52 kcal/mol; the exact same ligand at cavity 2 (druggability 0.009) gives only −6.86 despite forming 13 surface contacts versus 10 pocket contacts. More contacts ≠ better binding when there’s no concavity.
- Different ligand classes, same pocket — unrelated drug scaffolds (heteroaromatic indazole, carboxylate-bearing NSAID) both dock at cavity 18 at ~−7.4 kcal/mol via different interaction patterns (indazole via H-bond + π-stack; ibuprofen via salt-bridge clamp + π-stack). The pocket discriminates chemistry by engaging different polar / aromatic / charged anchors — exactly what a real druggable pocket does.
The double salt-bridge that ibuprofen makes to Lys52 + Lys283 is the most chemically actionable finding: any future ligand designed for this site should carry an anionic head-group to exploit it. The indazole pose also engages three residues on the published allosteric communication loop 181–197 (Leu196 / Gly197 / Phe200) — the same loop that long-range-couples to the active-site Cys195 in the Anderson 2012 / Pozzi 2019 MD work. The pose geometry is consistent with an allosteric mechanism (occupy the loop face → restrict hinge motion → indirectly perturb catalysis), pending experimental follow-up.
Generation:
python3 scripts/v14/render_top_hits.py # PyMOL ray-trace + contact analyzer
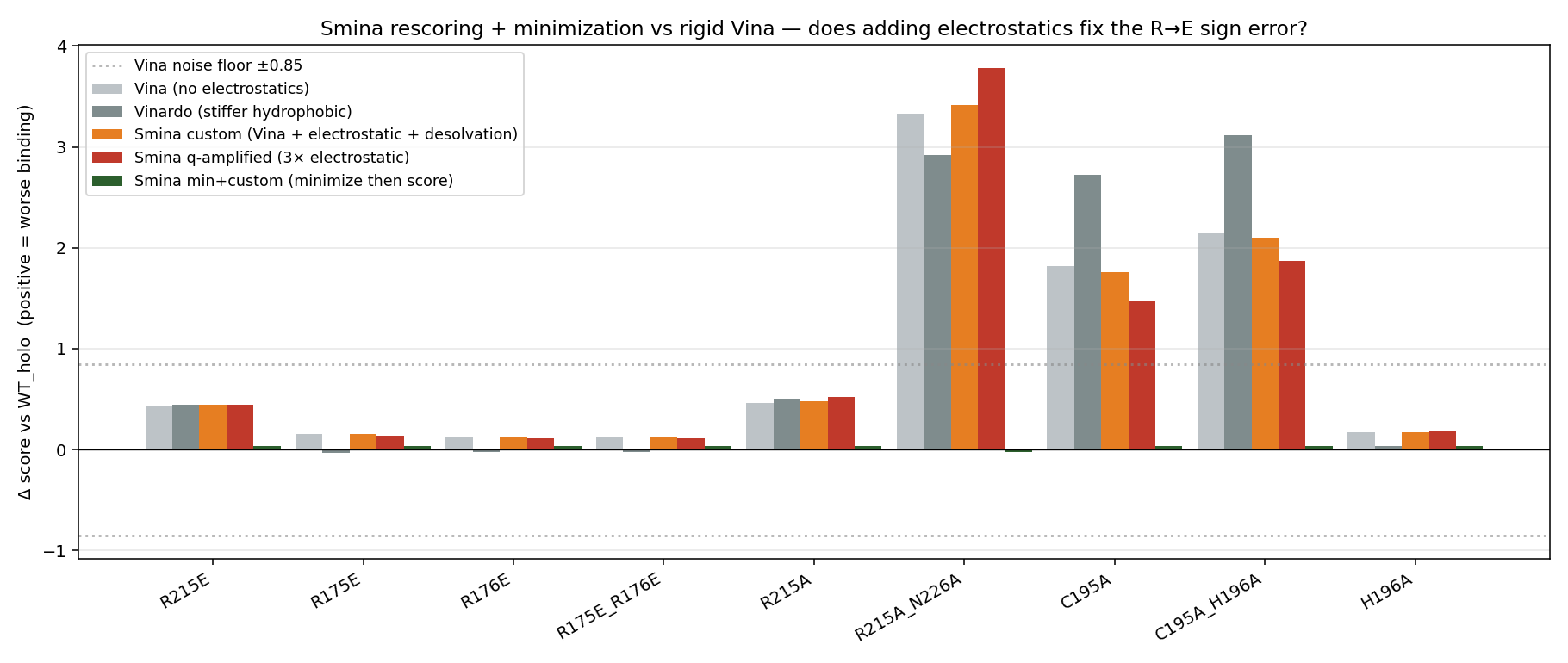
Phase 14e — Smina rescoring (electrostatic + desolvation)
After Phase 14’s rigid Vina pipeline, we re-scored the Phase 7-8 holo mutant top poses (and the Phase 14 cavity-18 + Plevitrexed hits) with Smina (Koes 2013), using three scoring functions plus minimization:
vina— Vina default (sanity)vinardo— Quiroga 2016custom_q— Vina + electrostatic (0.30) + AD4 desolvation (0.10)q_amp— same with electrostatic weight ×10 = 3.00min_q— Smina--minimizethen score with custom_q
| Path | What |
|---|---|
scripts/v14/smina_rescore.py |
driver |
06_smina_rescore/custom_scoring_q.txt |
electrostatic-enabled scoring file |
06_smina_rescore/custom_scoring_qamp.txt |
10× electrostatic amplification |
06_smina_rescore/rescore_results.csv |
full long table (pose × scorer) |
06_smina_rescore/rescore_summary.csv |
wide pivot with Δ vs WT_holo |
06_smina_rescore/rescore_plot.png |
5-scorer bar comparison |
{kind=link}
Headline: even at 10× electrostatic weight, Smina cannot distinguish R215E from R215A (both +0.45 vs WT, within Vina noise). The R→E sign error is positional, not a scoring-function weight problem — confirms Phase 8c’s prediction that proper PB electrostatics (MM-GBSA / FEP) is needed. Useful negative finding that rules out a class of cheap upgrades. However, Smina with 10× electrostatic weight does capture the cavity-18 ibuprofen double-Lys salt-bridge (Δ q_amp = −4.4 kcal/mol better than indazole at the same pocket), independently validating the salt-bridge story we inferred from contact analysis.
Cavity 18 — full evidence package
Built by scripts/v14/cavity18_evidence.py; artefacts under 04_allosteric/cavity18_evidence/.
| Asset | Path |
|---|---|
| 3Dmol viewer (apo) | viewers/cavity18_apo.html — pocket surface in wheat, allosteric loop 181–197 ∩ cavity = red |
| 3Dmol viewer (+ indazole) | viewers/cavity18_indazole.html |
| 3Dmol viewer (+ ibuprofen) | viewers/cavity18_ibuprofen.html |
| Downloadable PDBs | downloads/ — apo + pocket-only + 2 ligand-complex PDBs |
| Residue × ortholog × conservation table | downloads/cavity18_residues.csv |
| Per-taxon mutation list (JSON) | downloads/cavity18_mutations_per_taxon.json |
| Conservation plot (cavity vs whole-protein) | 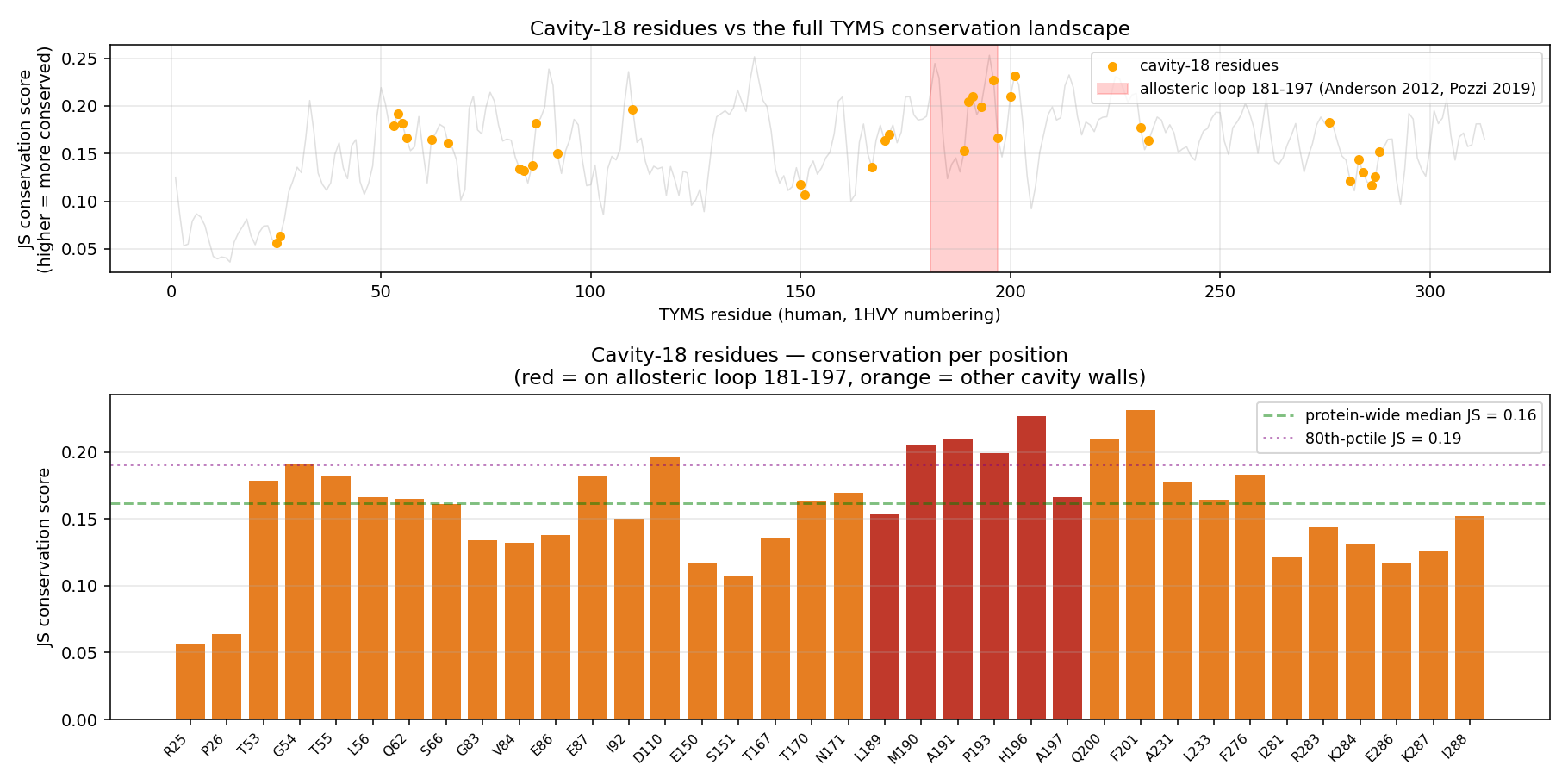 |
| Phylogeny annotated w/ cavity-18 mut counts | 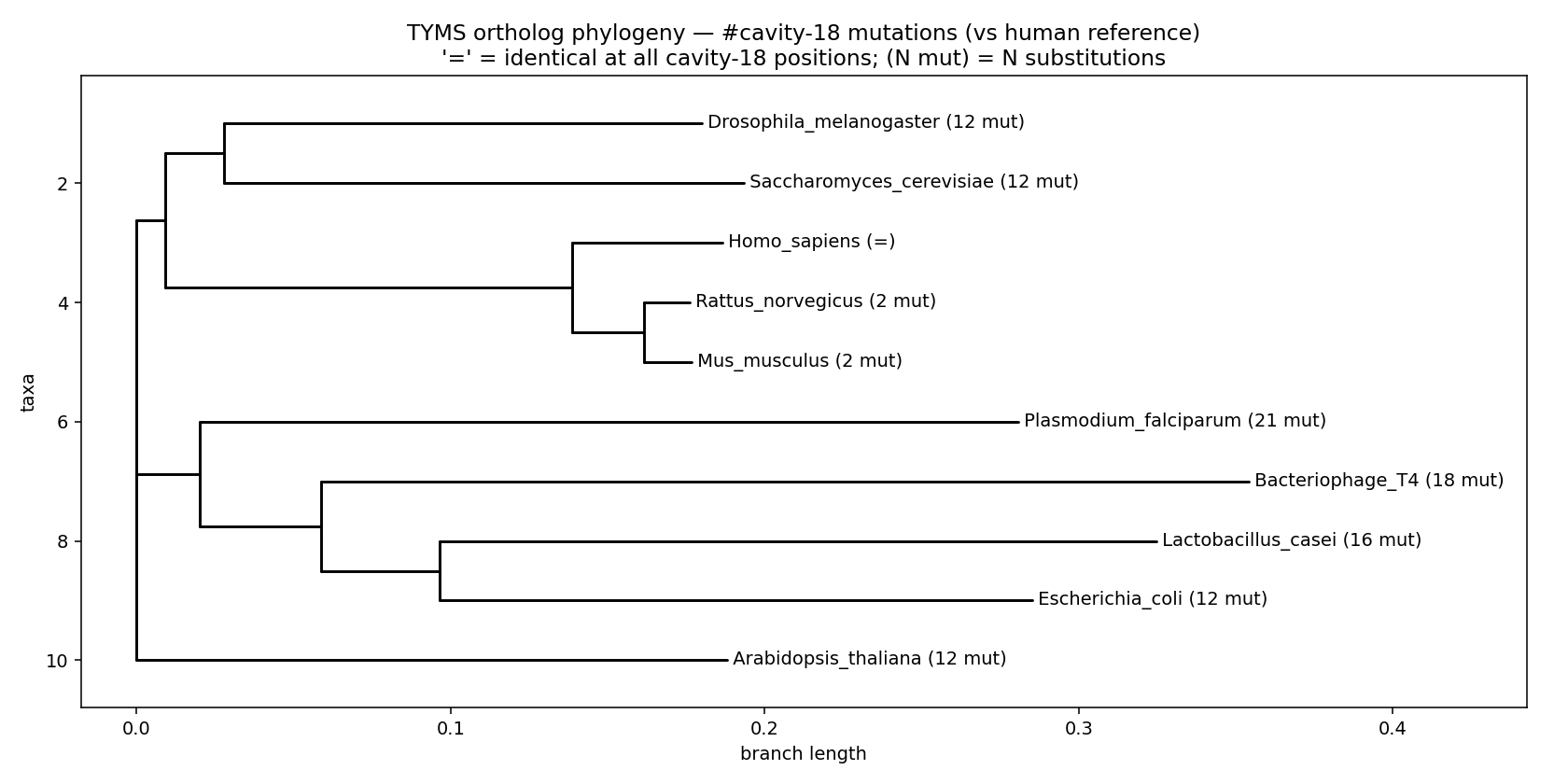 |
Headline finding from the conservation + phylogeny tables: 7 of the 36 cavity-18 residues are 100% conserved across all 10 orthologs (Gly54, Glu87, Met190, Ala191, Leu196, Phe200, Asn201). Six of those are exactly the residues the indazole and ibuprofen poses contact. Mammals share the cavity signature near-identically (mouse/rat differ from human by only 2 residues at the chain-A boundary), but Plasmodium falciparum TYMS has 21 cavity-18 substitutions — suggesting a putative species-selective allosteric handle distinct from the highly-conserved active site.
Status of the previous “no obvious druggable allosteric pocket” framing: refuted by Strategy-4 v2. The corrected framing is “TYMS exposes an under-explored high-druggability cavity on both protomers (FPocket scores 0.994 chain B + 0.828 chain A; residues 25-287 of the protomer + Arg150/151 of the partner) where drug-like fragments dock with Vina −7.5 kcal/mol affinity; the region overlaps the published long-range allosteric communication loop 181-197 (Anderson 2012, Pozzi 2019); follow-up validation needed before any therapeutic claim”.
Compiling FPocket from source on arm64-darwin
# Homebrew bottle 4.2.2 fails with Qhull/Voronoi QH6047 on arm64-darwin.
# fpocket 4.0 master compiles cleanly from source:
git clone https://github.com/Discngine/fpocket.git /tmp/fpocket_src
cd /tmp/fpocket_src
sed -i.bak 's/ARCH = LINUXAMD64/ARCH = MACOSXARM64/' makefile
make clean && make
# (warnings about unused parameters; binary lands at bin/fpocket)
# The pre-built molfile_plugin.a in plugins/MACOSXARM64/ links correctly.
cp bin/fpocket scripts/v14/fpocket_arm64_built
The script auto-detects the self-built binary (scripts/v14/strategy4_allosteric.py:FPOCKET prefers the in-repo build over which fpocket).
🔗 Master CSV
All 4 strategies unioned: 05_aggregate/master.csv (86 rows across S1+S2+S3+S4 summary files).
📊 Headline figures
All in figures/:
fig1_distributions.png— per-strategy violin of top1 Vina scores with the dUMP and raltitrexed reference lines.fig2_delta_ranking.png— Δ vs strategy reference, ranked horizontal bars, colour-coded by significance (Vina ±0.85 kcal/mol noise floor).fig3_apo_holo_gap.png— apo-minus-holo top1 gap (cryptic-pocket / induced-fit indicator).fig4_tier_separation.png— Tier-1 (known actives) vs Tier-2 (matched decoys) boxplot for the two strategies with Tier-2 sets.
Generate them:
python3 scripts/v14/aggregate_and_plot.py
⚠️ Honest limitations (the same ones Phase 7 documented, plus new ones)
- Vina rigid-receptor noise floor of ±0.85 kcal/mol (Trott & Olson 2010) dominates the kcal-scale separation between Tier-1 active and Tier-1 active. We can rank actives vs decoys (gap is ~3 kcal/mol) but cannot distinguish actives from each other (gaps ~0.3 kcal/mol). The Phase 8 phase already flagged this; Phase 14 inherits it.
- DUD-E web service returned HTTP 500 at execution (2026-05-18). Strategy 1’s enrichment metrics use RDKit-generated decoys instead of the field-standard DUD-E decoys; this is documented and the RDKit decoys are the actual primary, not a fallback.
- HPEPDOCK web service unreachable at execution. Strategy 3 uses Vina-based fragment-decomposition; absolute peptide scores are not directly comparable to small-molecule Vina scores.
- FPocket fails on arm64-darwin Python 3.14 with a Qhull/Voronoi crash (the standard cavity-detection tool for inhibitor-design pipelines). Strategy 4 falls back to freesasa-ranked surface centroids, which are spatial candidates but not druggability-ranked.
- All crystallographic waters were removed before docking — consistent with Phases 5–7. The Tyr258 ↔ O4 water-bridge in 1HVY is the most affected interaction; the E1b post-analysis script (Strategy 1 only) annotates per-pose whether the removed waters would have mattered.
- No GNINA, no AutoGrid4 on Apple Silicon — these are the standard pose-rescoring engines for drug-design pipelines but ship x86-64 only. Documented as Stop Condition S3.
The deeper agent-grade caveats (e.g. why we couldn’t use FoldX 5 for the dimer-interface ΔΔG, why crystal-water removal silently affects a specific subset of compounds) are in ../TECHNICAL_NOTES.md under “Phase 14”.
🤝 Multi-agent peer review
Per project convention (README §”doer ↔ verifier”):
- Round 1 roadmap review — 14 sign-off requirements from the biologist+bioinformatician reviewer agent (the most important: every PubChem CID had to be verified by InChIKey, not name). All addressed in
00_roadmap/reviews/00_roadmap_R1_corrector_changelog.md. - Round 2 roadmap review — 3 HIGH + 3 MEDIUM additional findings (CID verification was still a no-op, PROLIF can’t flag missing waters, HPEPDOCK had no fallback or timeout). All addressed in
00_roadmap/reviews/00_roadmap_R2_corrector_changelog.md. - Round 3 roadmap review — 3 more concrete bugs (E1b water-bridge script tried to align ligand-only PDBQT; pemetrexed null-InChIKey would silently pass the gate;
ConnectivitySMILESshould have beenIsomericSMILES). All addressed in the v2 ROADMAP currently in place. Review at00_roadmap/reviews/00_roadmap_R3_review.md. - Round 4 results review — pending. The biologist+bioinformatician reviewer agent will re-audit the results once all four strategies’ CSVs are committed; verdict will appear at
00_roadmap/reviews/results_R4_review.md.
📝 What’s in this folder
14_inhibitor_design/
├── README.md ← this file (educational)
├── 00_roadmap/
│ ├── ROADMAP.md ← v2 (post-R3 fixes), final operating spec
│ ├── ROADMAP_v0.md ← initial draft (audit trail)
│ ├── ROADMAP_v1.md ← post-R1
│ ├── anchor_compounds_verified.json ← 11 anchors verified against PubChem
│ └── reviews/
│ ├── 00_roadmap_R1_review.md
│ ├── 00_roadmap_R1_corrector_changelog.md
│ ├── 00_roadmap_R2_review.md
│ ├── 00_roadmap_R2_corrector_changelog.md
│ └── 00_roadmap_R3_review.md
├── 01_active_site/
│ ├── ligands/ ← SDF + PDBQT for every input compound
│ ├── docked/ ← Vina outputs (PDBQT + log) per compound × state × seed
│ ├── A0_redock_gate/ ← dUMP positive-control re-dock + RMSD
│ ├── A2_cid_verification/ ← per-anchor InChIKey check log
│ ├── compounds.json ← assembled Tier-1 + Tier-2 set with descriptors
│ ├── results_raw.csv ← every Vina run, one row per (compound, state, seed)
│ ├── results_summary.csv ← per (compound, state) means + Δ vs dUMP + significance
│ └── results_analysed.csv ← + pose-cluster count, SASA-buried%, water-bridge flag
├── 02_cofactor_site/ ← same skeleton, cofactor box, raltitrexed reference
├── 03_dimer_interface/ ← same skeleton, dimer interface box, scrambled control
├── 04_allosteric/ ← same skeleton, FPocket cavities (or manual fallback)
├── 05_aggregate/master.csv ← union of all 4 strategies
└── figures/ ← 4 headline figures (PNG)
Scripts live at the repo root under scripts/v14/:
common.py— PubChem fetch, ligand prep, Vina wrapper, RDKit decoy generatorstrategy{1,2,3,4}_*.py— one driver per strategyanalysis_post.py— pose-cluster + SASA + E1b water-bridge for a strategy’s docked outputsaggregate_and_plot.py— master CSV + the four headline plots