aminak
🧬 aminak — TYMS / dUMP structural-bioinformatics workbench
A teaching repository. An end-to-end pipeline that takes a single protein from a database accession through cross-species conservation, structure-based docking, mutational probing, and homology modelling — with independent multi-agent peer review at every step. The worked example is the molecular pair targeted by the colorectal-cancer chemotherapy drug 5-fluorouracil: human TYMS and its natural substrate dUMP.
📚 This file is the teaching face of the project. The agent-grade technical notes (audit history, build defects, what each iteration fixed and why) live in TECHNICAL_NOTES.md. The commit-by-commit story is in CHANGELOG.md. Reviewer reports are verbatim under
reviews/,reviews_v2/, …,reviews_phase6/.
📖 Quick glossary (read this first)
| Term | What it means in plain English |
|---|---|
| 🧪 TYMS | Thymidylate Synthase — the enzyme this project targets. UniProt accession P04818, a 313-residue protein that works as a homodimer (two copies stuck together). |
| 🧬 dUMP | 2′-deoxyuridine 5′-monophosphate — the natural substrate of TYMS. The molecule the enzyme normally binds and methylates to make dTMP, a DNA building block. |
| 💊 5-FU | 5-fluorouracil — chemotherapy drug used in colorectal cancer. It hijacks TYMS by being mistaken for dUMP. So a mutational probe of dUMP binding is, by construction, a probe of how 5-FU resistance arises. |
| 🧱 PDB 1HVY | The X-ray crystal structure we anchor on: human TYMS at 1.9 Å resolution, with dUMP and a folate-mimic cofactor (raltitrexed, residue D16) bound. |
| 🔍 Δ Vina score | The change in AutoDock Vina docking score when a residue is mutated. By convention here, positive = destabilising (mutant binds worse than WT). |
| 🔓 apo | Receptor with the cofactor pocket empty — only the protein, no raltitrexed (D16) bound. We dock dUMP into this state to ask “what would dUMP do if it arrived first?”. The whole pocket is available, so Vina finds tighter geometric fits → more negative scores. |
| 🔒 holo | Receptor with the cofactor pre-positioned in the folate-binding sub-pocket (the way the 1HVY crystal was captured). We dock dUMP into this state to ask “what does dUMP do once cofactor is already bound?”. The cofactor occupies part of the box, dUMP has less room → less negative scores in rigid-receptor Vina. Both states are reported because the biological binding order in TYMS is dUMP-first then cofactor-second; the holo dock is the inverted-order question. |
| 🧠 Modeller | A homology-modelling package: given an amino-acid sequence and known similar 3-D structures, it predicts a 3-D model of the unknown protein. |
| 🤝 doer ↔ verifier | The audit pattern this repo follows: a “doer” agent runs the pipeline; four specialised “verifier” agents (validator / code reviewer / scientific officer / structural bioinformatician) independently audit; the doer fixes; repeat until clean. |
🧠 Why TYMS — what it is and why it matters
Thymidylate synthase (TYMS · EC 2.1.1.45 · UniProt P04818 · PDB 1HVY) catalyses the only de-novo route to thymidylate: dUMP + 5,10-methylene-THF → dTMP + DHF. It is the rate-limiting enzyme for DNA synthesis — block TYMS and dividing cells stall in S phase. That makes the human enzyme a 60-year-old validated anticancer target: 5-fluorouracil, capecitabine, raltitrexed, pemetrexed, and plevitrexed all converge on its active or cofactor pocket, and combined annual prescribing exceeds two million patients globally (colorectal, gastric, breast, lung, mesothelioma).
The same enzyme matters in malaria. Plasmodium falciparum carries a bifunctional DHFR-TS fusion in which the C-terminal half is a thymidylate synthase; antifolates like pyrimethamine inhibit the DHFR domain, but resistance is widespread and selective inhibition of the parasite TS domain itself has been a long-running medicinal-chemistry goal. The catch is that the active site is conserved between species (so 5-FU works in both, with no selectivity), and the selectivity handle has to come from somewhere outside the conserved core — the central premise of this project’s Strategy 4.
TYMS is also a tractable structural target: a 313-aa, mostly α/β fold with an obligate homodimer interface, a deep cofactor pocket, and a flexible 181–197 loop that long-range-couples to the catalytic Cys195 (Anderson 2012, Pozzi 2019). Structural enrichment of new chemical matter — induced-fit pockets, conserved-vs-divergent residues, fragment-level binding — is therefore the natural way to ask: where else on TYMS could a small molecule bind, and which of those sites are species-selective?
| Why we chose this target | Rate-limiting in dTMP biosynthesis · validated antitumour + antimalarial · published 313-aa homodimer · multiple FDA-approved inhibitors as positive controls |
| What hasn’t been done well | Selectivity between Plasmodium and human TS, and any non-orthosteric (cryptic / allosteric) drug-binding site on the human enzyme |
| What this project asks | (1) Can rigid Vina see active-site mutations? (null) (2) Where else on TYMS is there a druggable pocket? (cavity 18) (3) Is that pocket structurally divergent enough between Pf and Hs to support a species-selective lead? (next two sections) |
🎯 What this project actually does, in one paragraph
We take human TYMS (P04818), align it to ≥ 10 verified orthologs from across the tree of life to find conserved residues, intersect those with database-annotated active-site residues, dock the natural substrate dUMP into the chain-A active site of the experimental crystal structure (PDB 1HVY) under two cofactor conditions, then build a panel of 20 single and double mutants at active-site residues and re-dock under both conditions. We then ask: can a rigid-receptor docking pipeline distinguish these mutants by their Δ Vina score? Separately, in Phase 6, we use Modeller to build 10 homology models of TYMS from BLAST-discovered templates at 30–95 % sequence identity (deliberately educational, not 100 %) and validate them with Ramachandran φ/ψ + DOPE profiles + RMSD vs the experimental crystal.
💡 The teaching point
Rigid-receptor AutoDock Vina with AD4 partial charges and the physically correct (net −2) raltitrexed cofactor cannot resolve TYMS active-site point mutants at the kcal/mol scale. Across 20 mutants × 2 cofactor conditions, the largest holo Δ Vina score is +0.77 kcal/mol (R215A_N226A) — well below Vina’s documented noise floor of ±0.85 kcal/mol (Trott & Olson 2010; Forli et al. 2016). The ranking is directionally chemically sensible (R215 phosphate clamp, H196 catalytic dyad, N226 substrate orientation) but statistically silent.
This is reported as a null-result methodology paper — and that itself is a teaching point: an honest null result, with the limitation owned, is more useful than an unhonestly-positive one. The next step (induced-fit / RosettaDock / GNINA scoring) is signposted but not executed.
Phase 14 update. A subsequent inhibitor-design phase asked: forget mutants — what molecules will out-compete dUMP, or bind elsewhere on the enzyme? Four binding sites screened in parallel (active-site dUMP-mimetics, cofactor-site antifolates, dimer-interface PPI disruptors, allosteric / surface hotspots), three reviewer/corrector rounds on the roadmap before any compute, three more rounds on the results. Two findings — both honestly bounded:
(a) Plevitrexed (ZD9331) reproduces as the only above-noise hit in the cofactor pocket — top1 −10.01 kcal/mol, Δ −0.88 vs raltitrexed. This is 0.03 kcal/mol above the ±0.85 kcal/mol Vina noise floor — at the noise floor, not comfortably above it. Reproducible across both seeds and consistent with the published sub-nanomolar TYMS Ki (Jackman 1997); read as a positive control passing, not a de novo discovery.
(b) A geometrically druggable third pocket on the TYMS surface — FPocket druggability score 0.994 (with C2-symmetric mirror at 0.828) overlapping the published allosteric communication loop 181–197 (Anderson 2012, Pozzi 2019). Two promiscuous drug-like fragments (1H-indazole, ibuprofen — both known off-target binders of many proteins) dock at −7.5 kcal/mol. This is a geometry + conservation hypothesis, not validated affinity — a real follow-up needs specific ligands and either ITC/SPR or a TYMS-specific MD ensemble; the bare docking of promiscuous fragments is necessary evidence (the pocket can be filled) but not sufficient evidence (no selectivity demonstrated). Full write-up in § Phase 14 below.
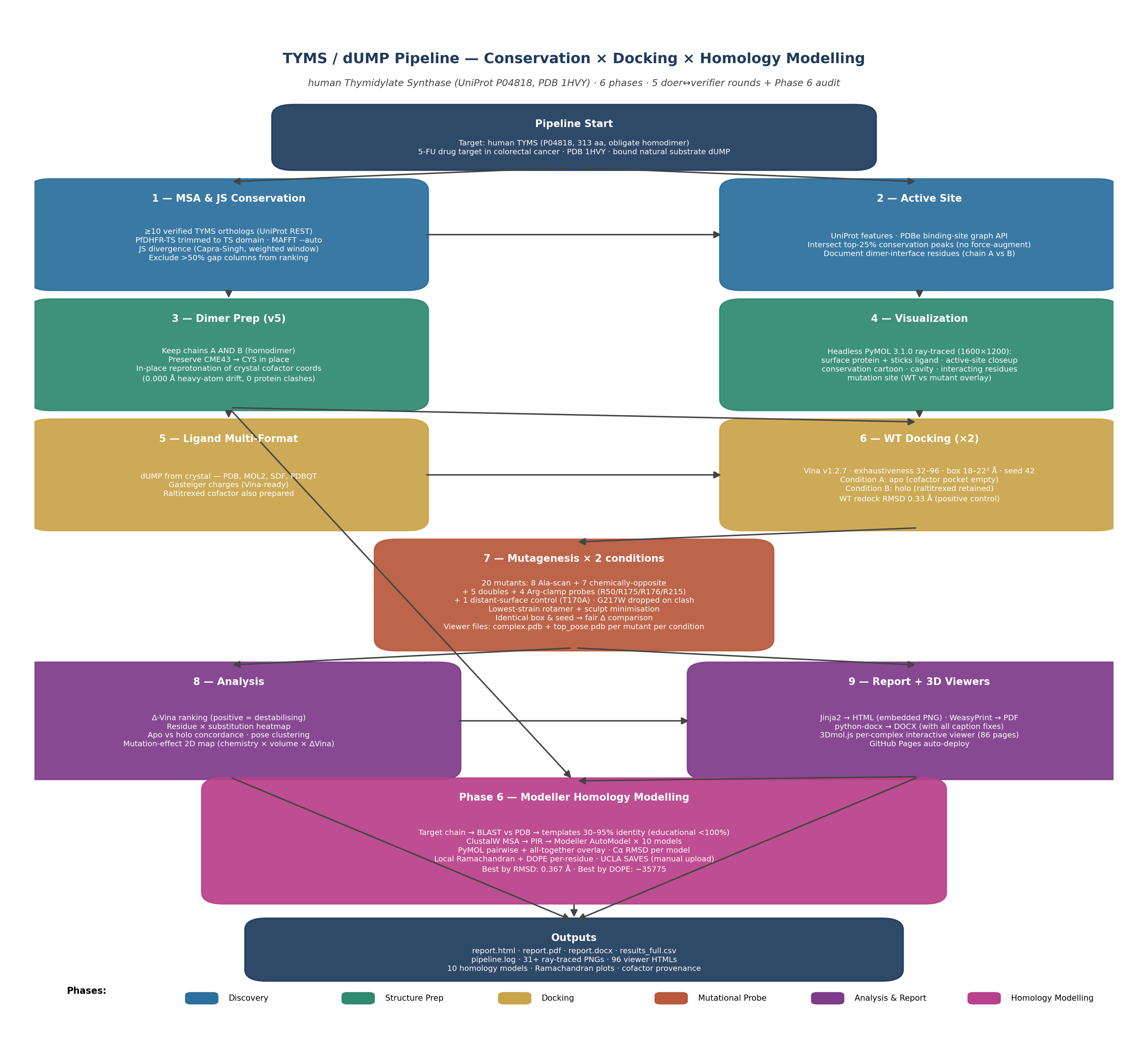
🧭 Pipeline at a glance
Eight phases, nine stages each in its own numbered subfolder. The full Mermaid diagram is concise on purpose; click any phase to jump to the relevant section.
flowchart LR
classDef d fill:#2b6f9c,stroke:#1f4f70,color:#fff
classDef p fill:#2f8a6f,stroke:#1f6452,color:#fff
classDef k fill:#caa44a,stroke:#a4842c,color:#1a1a1a
classDef m fill:#b8593c,stroke:#8a4128,color:#fff
classDef r fill:#7e3b8a,stroke:#5d2864,color:#fff
classDef h fill:#b9408a,stroke:#8a2b66,color:#fff
classDef i fill:#3a5a8c,stroke:#1f3a64,color:#fff
S1[1 · MSA + JS conservation]:::d
S2[2 · Active site]:::d
S3[3 · Dimer + cofactor prep]:::p
S4[4 · PyMOL renders]:::p
S5[5 · Ligand multi-format]:::k
S6[6 · WT docking - apo and holo]:::k
S7[7 · Mutagenesis ×20 ×2 conditions]:::m
S8[8 · Analysis]:::r
S9[9 · Reports + 3Dmol viewers]:::r
P6[Phase 6 · Modeller homology modelling]:::h
P7[Phase 7 · Multi-replica / SASA / AlphaFold / phylogeny]:::m
P8[Phase 8 · Vinardo + flex-residue Vina]:::r
P14[Phase 14 · Inhibitor design at four sites]:::i
S1 --> S2 --> S3 --> S5 --> S6 --> S7 --> S8 --> S9
S3 --> S4 --> S6
S3 --> P6
S9 --> P6
S9 --> P7 --> P8 --> P14
Full-detail static diagram: workflow_diagram_v3.png.
{kind=link}
🗺️ Repo map — clickable
Every coloured block below is a clickable link to that folder on GitHub. The diagram is committed as a placeholder SVG and is auto-refreshed by repo-visualizer (GitHub Next) on every push to main.

🔭 Live 3D structures
The protein–ligand complex on this page rotates in place; the click-through buttons open the same structure in a full interactive viewer (drag-rotate, scroll-zoom, surface-toggle, spin).
 WT (apo) + dUMP Wild-type TYMS docked with its natural substrate dUMP. Cofactor pocket left empty. ▶ 3Dmol viewer · ▶ Mol* viewer |
 WT (holo) + dUMP + cofactor Same WT receptor with the folate-mimic cofactor (raltitrexed) retained. Physiologically realistic. ▶ 3Dmol viewer · ▶ Mol* viewer |
 R215A_N226A holo — top destabiliser Double mutant: phosphate-clamp Arg215 → Ala and substrate-orienting Asn226 → Ala. Largest holo Δ Vina (+0.77). ▶ 3Dmol viewer · ▶ Mol* viewer |
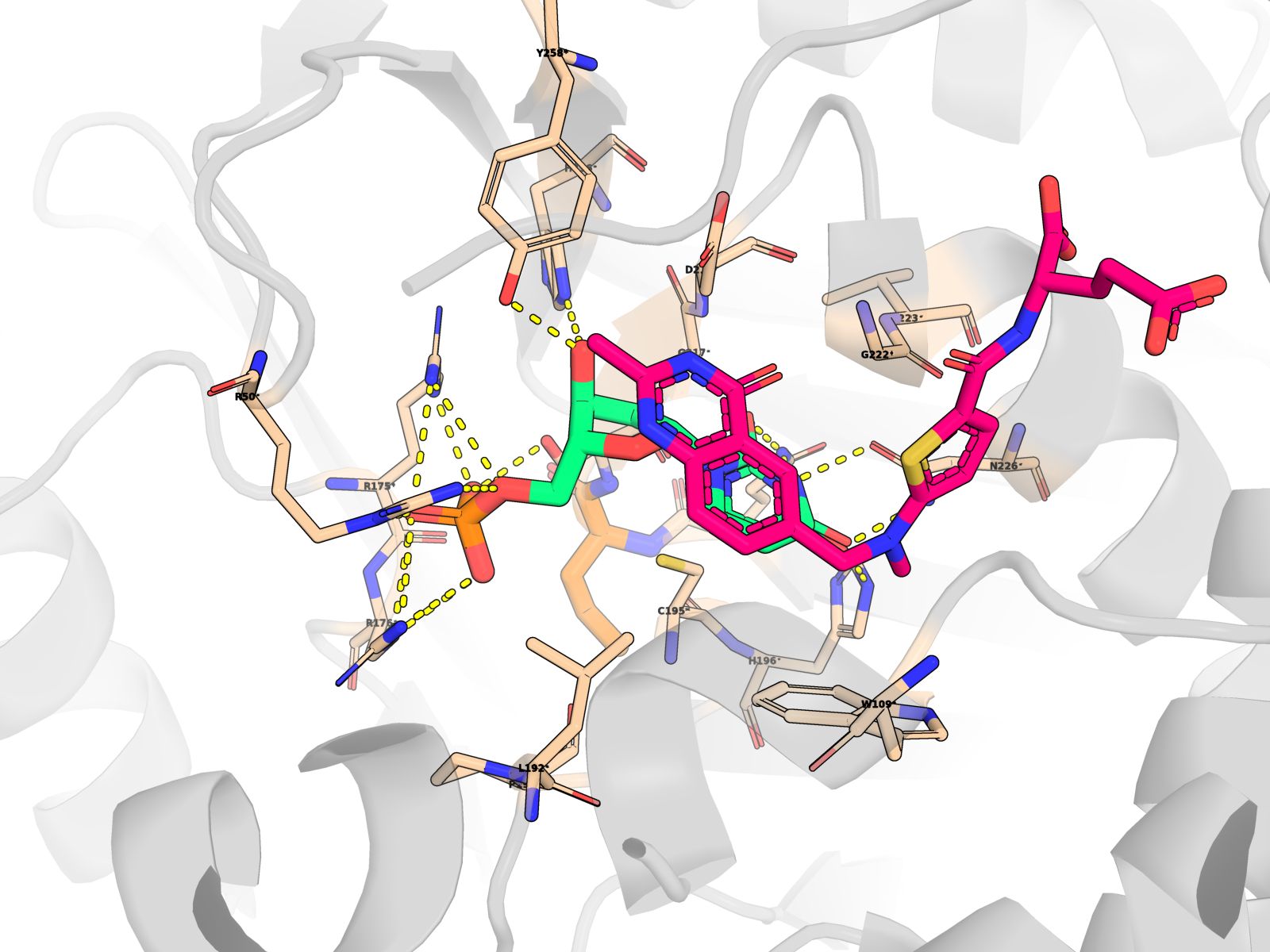
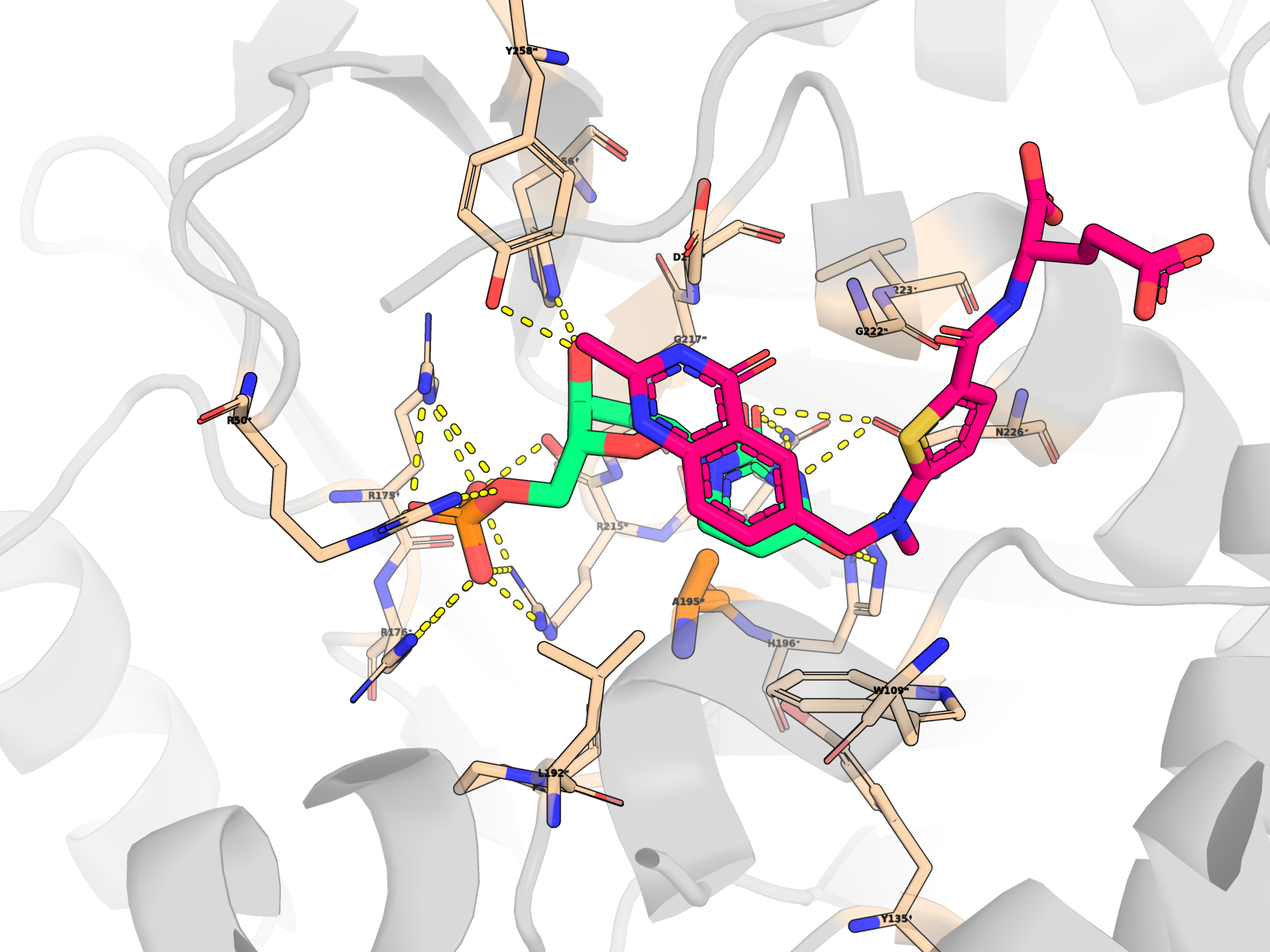
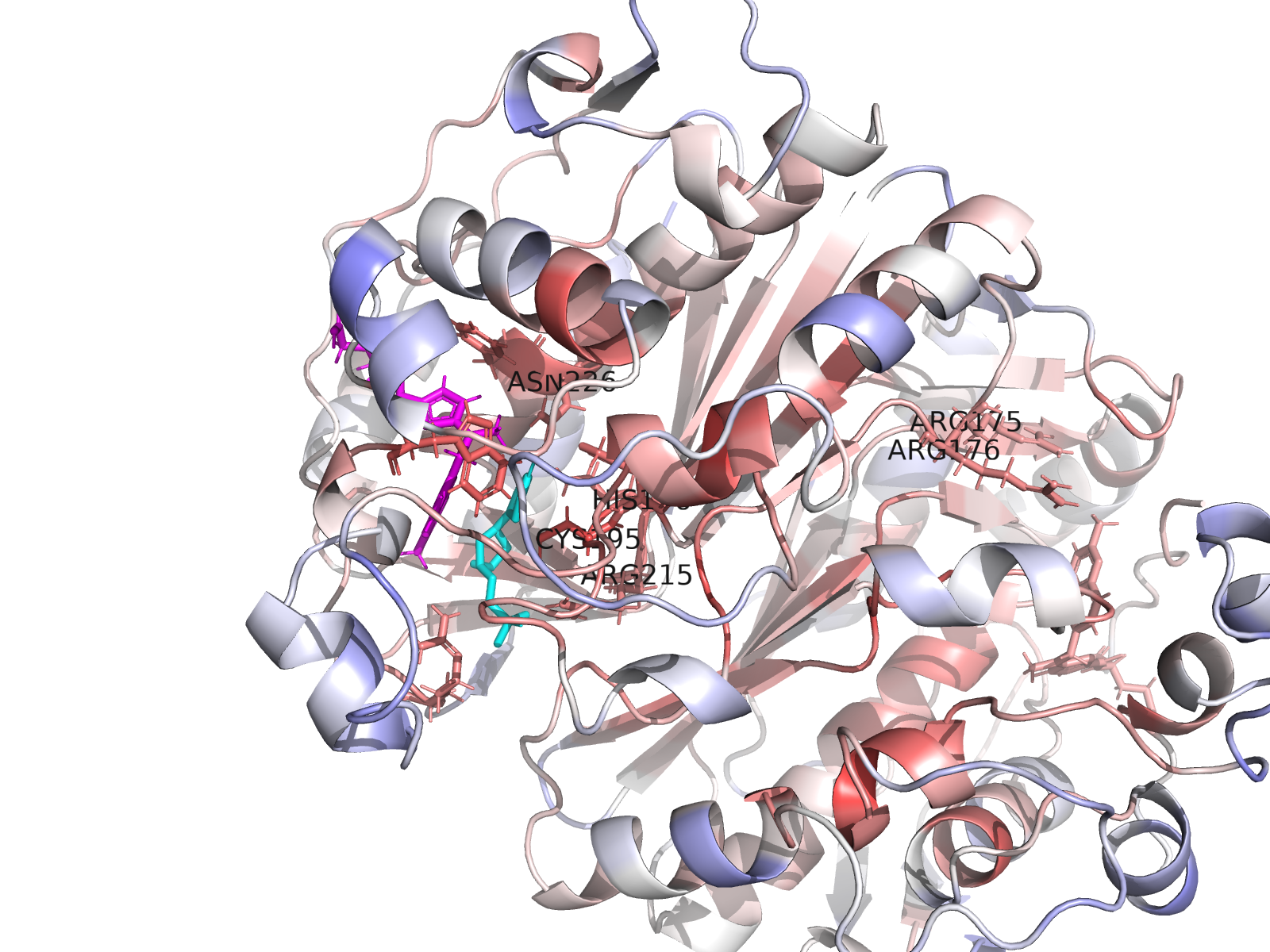
A complete index of 96 interactive viewer pages is at ariomoniri.github.io/aminak/viewers/. In every viewer the protein is rendered as cartoon + semi-transparent surface, the ligand as fat magenta sticks, active-site residues as labelled sticks; catalytic Cys195 / His196 / Arg175 / Arg176 / Arg215 / Asn226 carry permanent text labels.
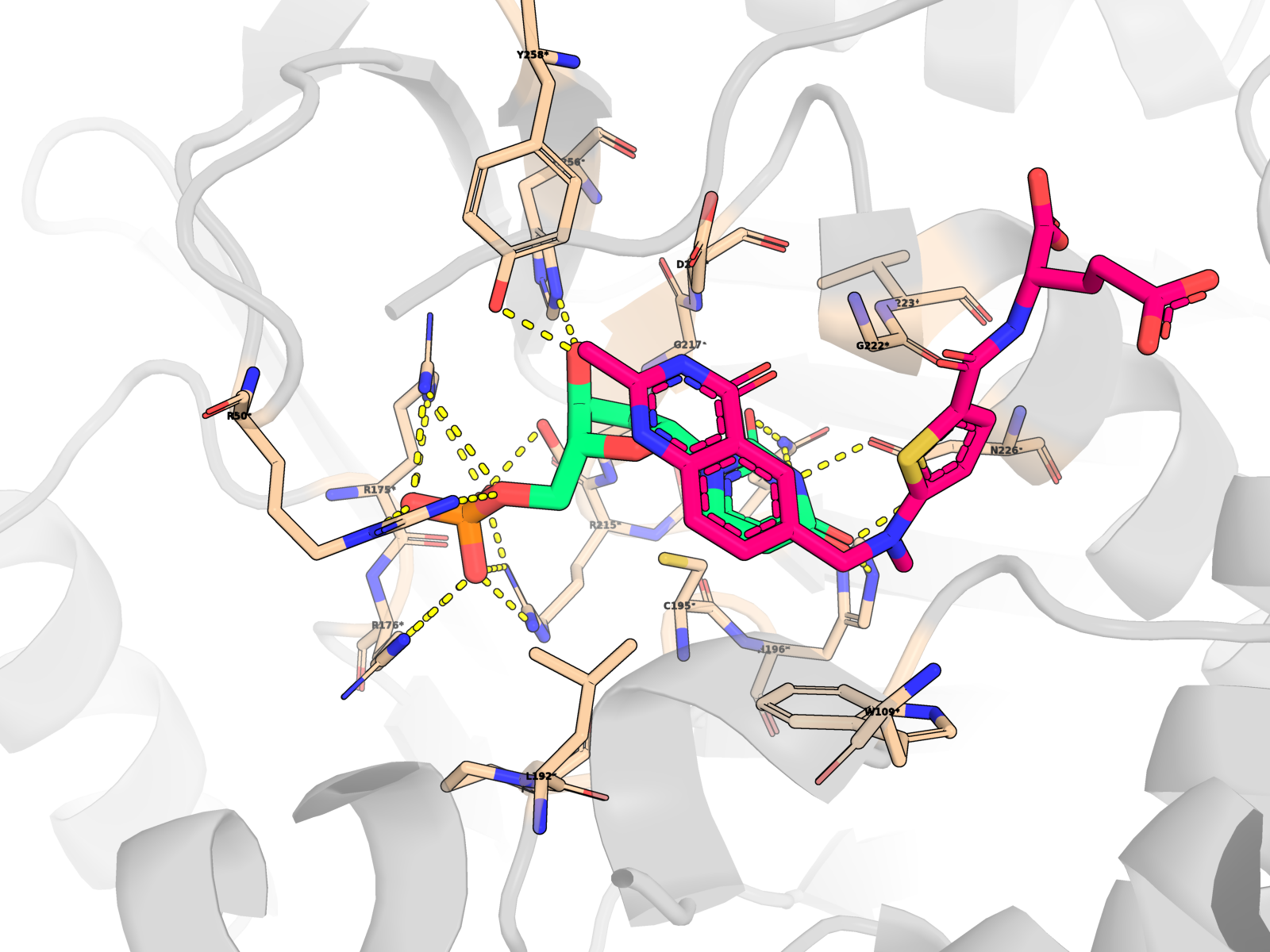
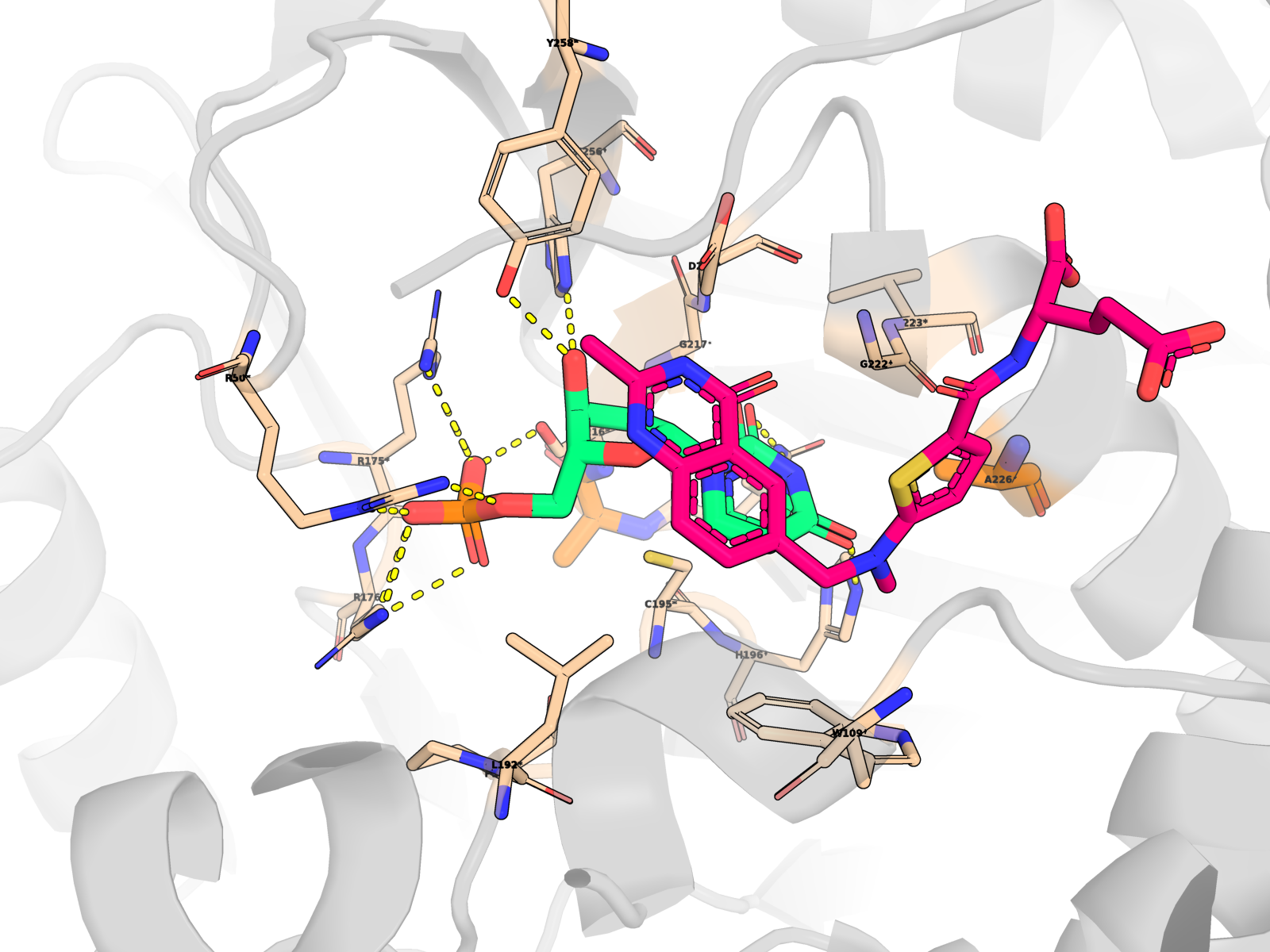
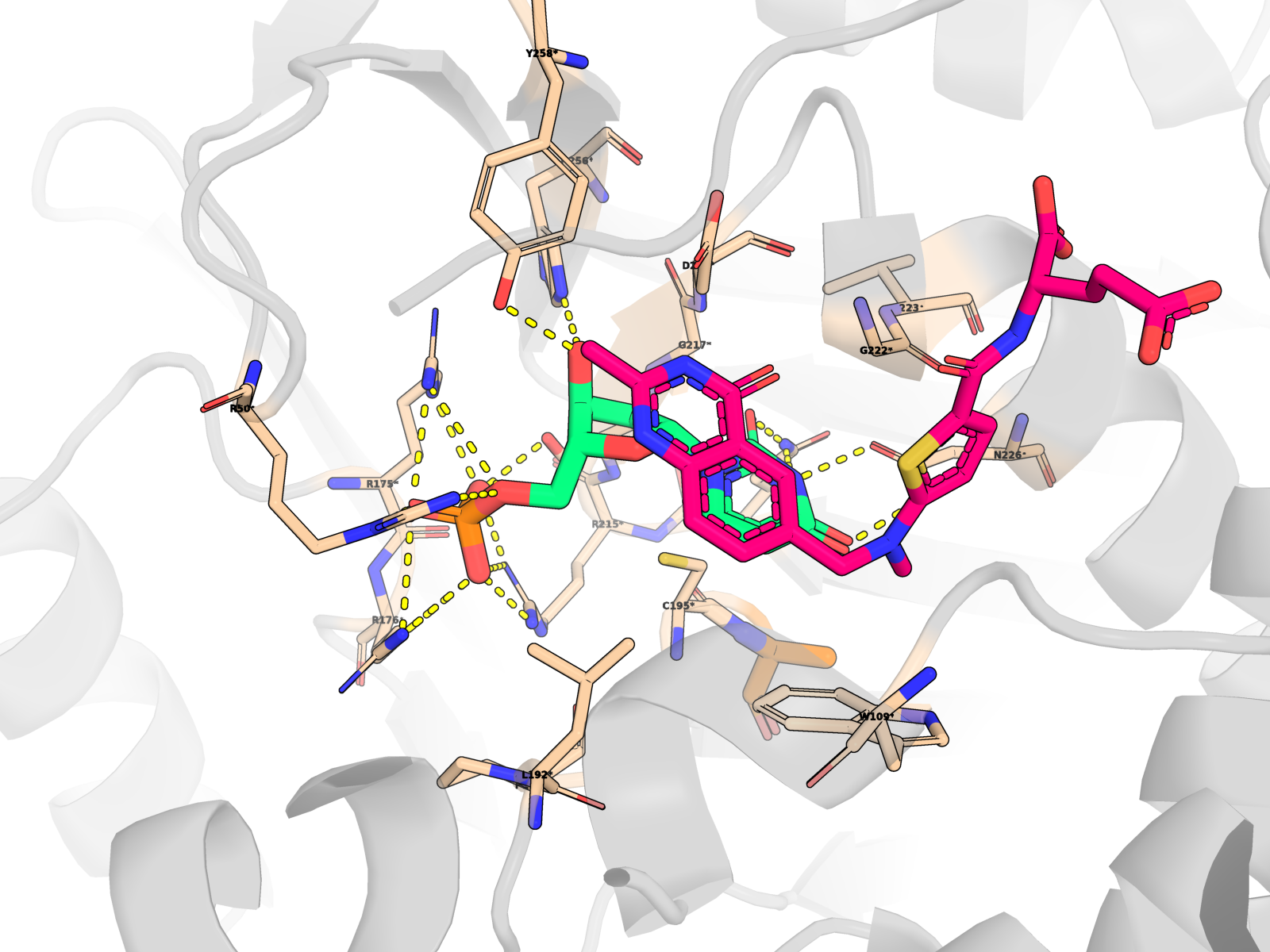
🖼 Publication-quality renders — close-ups of the active site
Cartoon protein (grey, semi-transparent) + active-site residues within 5 Å of dUMP as wheat sticks (every residue is labelled XNNN*; the asterisk is a uniform marker, not an interaction-only flag) + dUMP substrate in limegreen + cofactor raltitrexed (D16) in hotpink (clearly distinct from dUMP) + mutated residue in orange + dashed yellow lines for polar (N/O ↔ side-chain N/O) contacts < 3.5 Å only between dUMP and protein. All hydrogens hidden (heavy-atom-only renders) on white opaque ray-traced background.
About the second pink molecule: TYMS is an obligate homodimer with two equivalent active sites, so the holo PDB contains two raltitrexed cofactor copies (one per protomer, chains A + B) plus one docked dUMP pose (chain A active site). The renders zoom to the chain-A active site, so the chain-B cofactor is mostly out of frame; the in-frame pink molecule is the chain-A raltitrexed. This is the correct biological state, not a rendering artefact.
 |
 |
 |
| WT holo | R215A_N226A | H196A |
 |
 |
 |
| R215E | R50A | C195A |
All 7 renders (including R175E_R176E) in 12_phase7/07_pub_renders/, 1600 × 1200 ray-traced.
🌀 Note on the rotating GIFs above. The animated thumbnails show protein + dUMP only (cofactor is hidden) to keep the rotation visually clean. Earlier versions left raltitrexed cyan sticks visible, and the cofactor’s long polyglutamate tail extended past the protein surface, producing the “tentacles” / disrupted look. The static reference renders below (and the 3Dmol viewer click-throughs) show the full holo complex with all three ligands. See the “Why two ligands?” callout for the full ligand inventory.
❓ “Why do I see two ligands in the holo views?” Each holo complex has three ligand molecules, by design and consistent with the 1HVY crystal:
- dUMP (residue name
UMP, magenta sticks, chain X) — the substrate we docked. One copy, placed in the chain-A active site.- Raltitrexed (residue name
D16, cyan sticks) — the antifolate cofactor that occupies the methylene-THF pocket. Two copies, one per chain, because TYMS is an obligate homodimer and both subunits’ cofactor pockets are occupied in 1HVY.So a holo view shows 1 dUMP + 2 cofactors = 3 ligands total. The apo views show 1 dUMP and no cofactors. (In v5 we initially built
wt_holo_complex.pdbwithout the cofactors — that bug is now fixed, so the WT and mutant holo viewers are visually consistent.)
🖼 More structural views — click any thumbnail to open the live viewer
 H196A holo Catalytic dyad: His196 → Ala |
 R175E_R176E holo Phosphate-clamp charge inversion |
 T170A holo Distant-surface negative control |
 R215E holo Phosphate-clamp charge inversion (single) |
 R50A holo Phosphate-clamp bulk loss |
 C195A holo Catalytic Cys → Ala (flagged low-confidence) |
 Y258F_F225Y holo Aromatic-swap double mutant |
 Modeller model 3 Best by DOPE (Phase 6) |
 Modeller model 10 Best by Cα RMSD vs 1HVY |
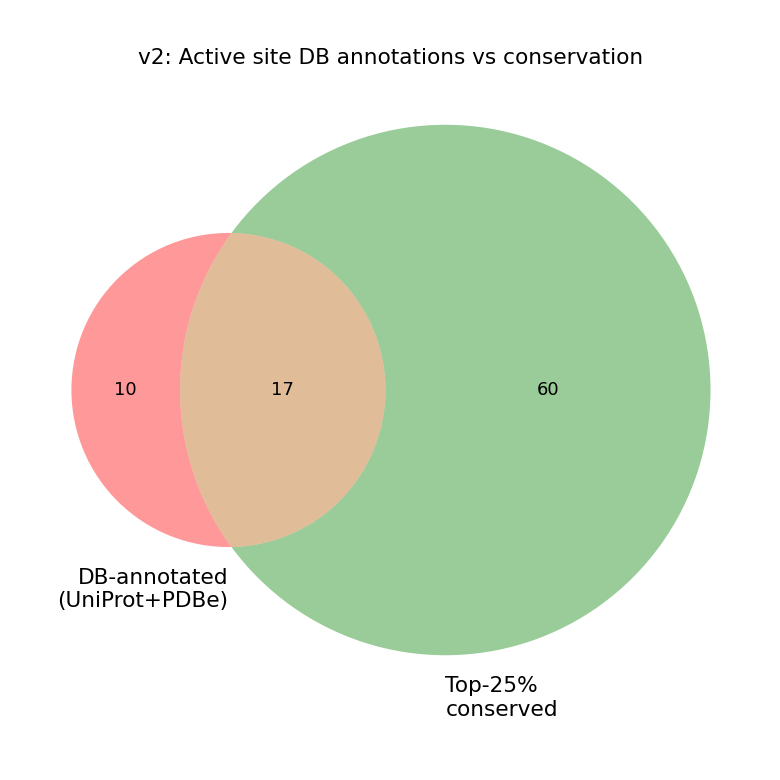
🔬 Stage 1 + 2 — Conservation and active-site annotation
We aligned the human TYMS sequence to 10 verified TYMS orthologs (UniProt REST) spanning Mus musculus, Rattus norvegicus, Escherichia coli, Lactobacillus casei, Saccharomyces cerevisiae, Drosophila, Arabidopsis, bacteriophage T4, and Plasmodium falciparum (DHFR-TS fusion, trimmed to its TS domain before alignment). Conservation was scored per residue with Jensen–Shannon divergence (Capra & Singh 2007, weighted window). Columns with > 50 % gap are excluded from percentile ranking — not just down-weighted.
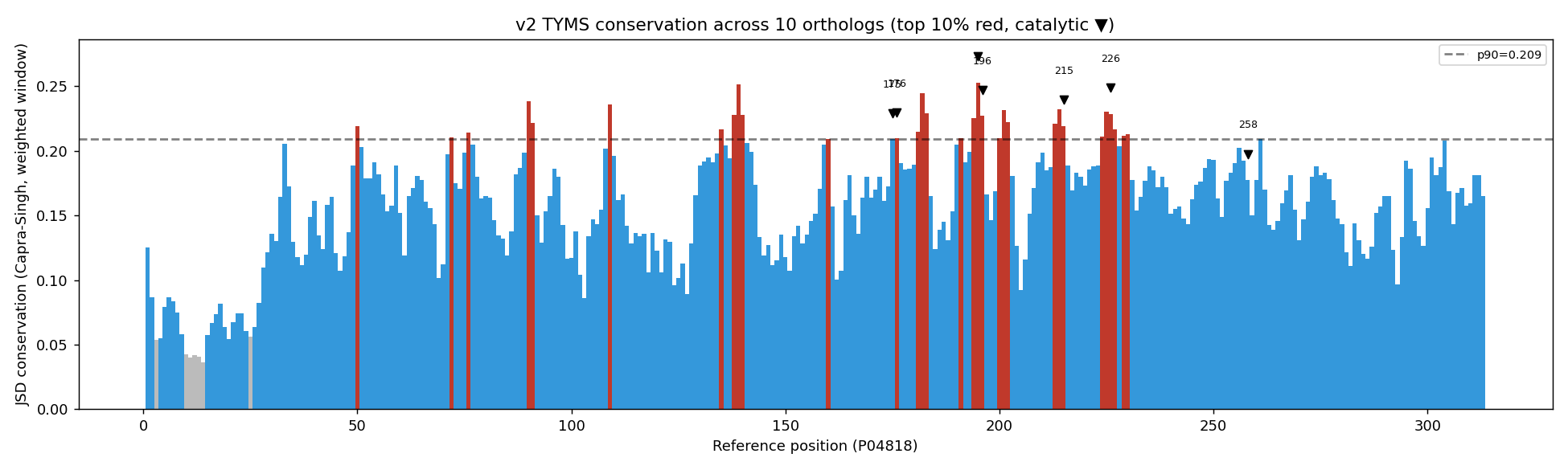 Top 10 % conserved positions are highlighted in red. The catalytic residues Cys195, His196, Arg175/176/215 and Asn226 all sit naturally in the top decile — no force-augmentation needed (an earlier version did need it; see CHANGELOG.md for the bug-fix story).
Top 10 % conserved positions are highlighted in red. The catalytic residues Cys195, His196, Arg175/176/215 and Asn226 all sit naturally in the top decile — no force-augmentation needed (an earlier version did need it; see CHANGELOG.md for the bug-fix story).
We then intersected the conserved residues with two database sources (UniProt features + PDBe binding-site graph API for 1HVY) to define the active-site set used in Stage 7.
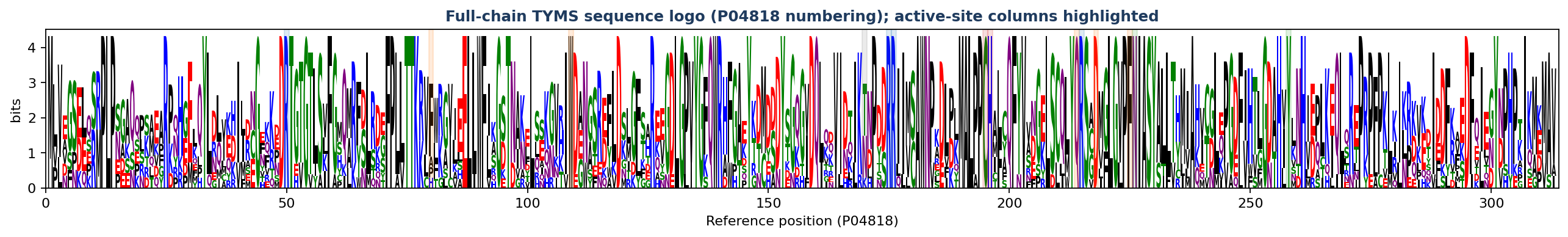
🔤 Sequence logo at the active-site & mutated residues
WebLogo-style stack at every residue we mutated. Letter height ∝ frequency × information content (bits = log₂20 − Shannon entropy across the 10 orthologs). A single tall letter spanning the full 4.32-bit ceiling means the residue is invariant across the panel; a stack of multiple shorter letters means the position is variable. Coloured band beneath each column = functional class.
![]()
What the logo tells you, position by position. The “Which species carry which residue?” column maps every non-WT amino acid back to the 10-ortholog phylogeny, so you can see which branches of the tree tolerate variation:
| Position | WT | Mutated to | Conservation across the 10 orthologs | Which species deviate from WT? | Class |
|---|---|---|---|---|---|
| R50 | R | →Ala, →Glu | R = 10/10 (100 %, invariant) | none | Phosphate clamp |
| F80 | F | →Ala, →Asp | F = 7/10 (70 %), H = 1/10, P = 1/10, A = 1/10 | E. coli = H · L. casei = P · phage T4 = A (all three bacterial / phage lineages) | Pocket scaffold |
| W109 | W | →Ala | W = 10/10 (100 %, invariant) | none | Pocket scaffold |
| T170 | T | →Ala | T = 5/10 (50 %), N = 4/10, K = 1/10 — variable, exactly as expected for the distant-surface control | E. coli, D. melanogaster, A. thaliana, P. falciparum all = N · phage T4 = K (the 5 mammalian / yeast / human-aligned lineages keep T) | Distant control |
| R175 | R | →Ala, →Glu | R = 10/10 (100 %, invariant) | none | Phosphate clamp |
| R176 | R | →Ala, →Glu | R = 10/10 (100 %, invariant) | none | Phosphate clamp |
| C195 | C | →Ala, →Ser | C = 10/10 (100 %, invariant) — the catalytic nucleophile | none | Catalytic |
| H196 | H | →Ala, →Phe | H = 10/10 (100 %, invariant) — the catalytic dyad partner | none | Catalytic |
| Q214 | Q | →Ala | Q = 10/10 (100 %, invariant) | none | Pocket scaffold |
| R215 | R | →Ala, →Glu | R = 10/10 (100 %, invariant) | none | Phosphate clamp |
| D218 | D | →Ala, →Lys | D = 10/10 (100 %, invariant) | none | Pocket scaffold |
| F225 | F | →Ala, →Asp | F = 10/10 (100 %, invariant) | none | Pocket scaffold |
| N226 | N | →Ala, →Asp | N = 10/10 (100 %, invariant) | none | Substrate orientation |
| Y258 | Y | →Ala, →Phe | Y = 10/10 (100 %, invariant) — substrate-orienting tyrosine | none | Substrate orientation |
The teaching point: 12 of the 14 active-site residues are 100 % conserved across all 10 orthologs (single tall letter on the logo), justifying every catalytic / phosphate-clamp / substrate-orientation choice as a meaningful probe. The two variable positions tell two different stories:
- F80 drifts only on the bacterial / phage branch (E. coli H, L. casei P, phage T4 A) — eukaryotic TYMS keeps F; the variation tracks a clean Bacteria-vs-Eukaryota split
- T170, the distant-surface control, drifts on every non-mammalian branch (T → N in E. coli / D. melanogaster / A. thaliana / P. falciparum; T → K in phage T4) — exactly the noise pattern you want to see for a residue that is functionally indifferent
Per-position frequency table (sorted, top-3 observed): 11_enhanced/aa_logo_active_site.csv. Full-chain sequence logo: 11_enhanced/aa_logo_full_chain.png (313 columns; active-site columns shaded by functional class).
{kind=link}
🌳 Why is the active site so conserved? — Phylogeny of the 10 TYMS orthologs
Distance-based neighbour-joining tree built from the v2 MSA with BLOSUM62 distances, with kingdom annotation per leaf. Teaching-grade only — no model selection, no bootstrap support. For a publication-grade topology, re-run with IQ-TREE / RAxML under LG+G or JTT+G+I with ≥ 1000 bootstraps.
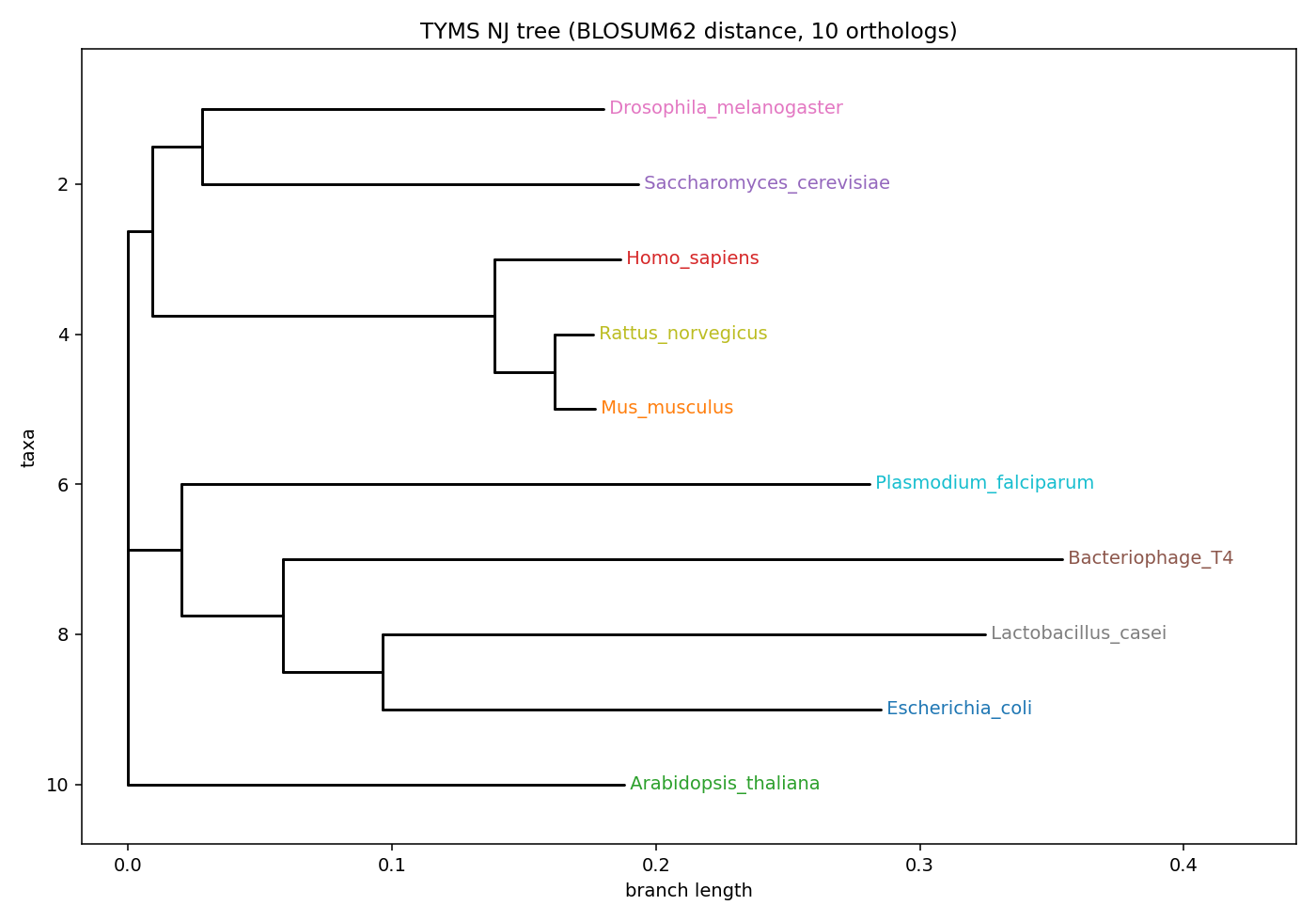
Newick + interactive HTML: 12_phase7/05_phylogeny/. The tree explains why the conservation logo above collapses to single tall letters at every catalytic / phosphate-clamp position — these orthologs span Metazoa, Plantae, Bacteria, Protozoa, and Bacteriophage, and the active-site chemistry is invariant across all of them. A residue conserved this widely is one that the protein cannot afford to lose — exactly the residues that should be most informative as mutational probes.
🧱 Stage 3 + 4 — Dimer-aware structure preparation and visualisation
TYMS works as an obligate homodimer with the active site spanning the chain-A / chain-B interface — the dUMP phosphate is clamped by Arg175′ and Arg176′ from the partner subunit. Stage 3 therefore keeps both chains, preserves the covalently-modified Cys43 (CME43) by re-mutating it back to native CYS in place, and re-protonates the bound cofactor without moving any heavy atom (0.000 Å heavy-atom drift, 0 protein clashes — verified).
 |
 |
|---|---|
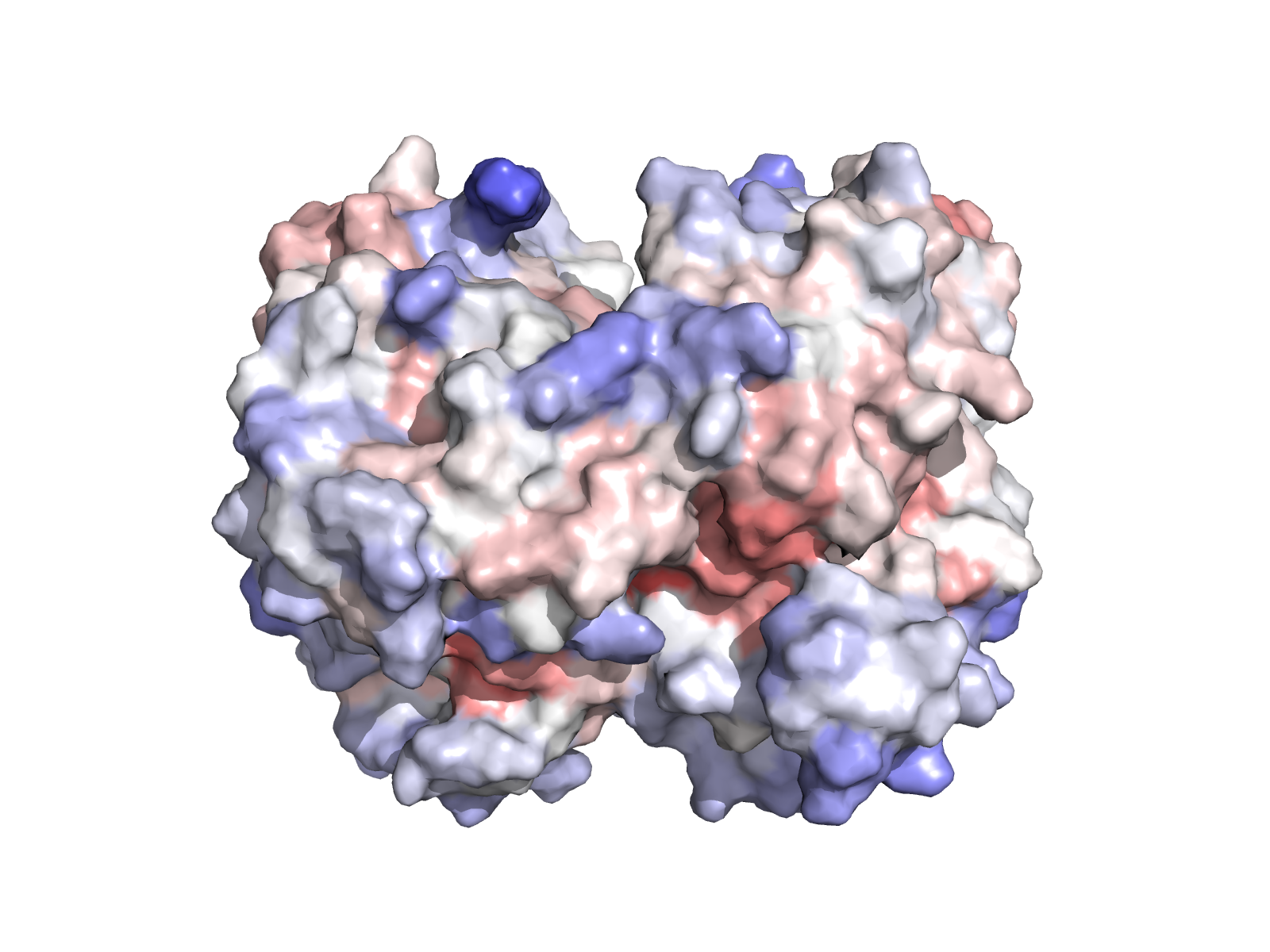
| TYMS homodimer (chains A + B). dUMP highlighted. | Surface coloured by Jensen–Shannon conservation. |
 |
 |
|---|---|
| Chain-A active-site closeup with residue labels. | Cys195 – His196 catalytic dyad geometry. |
🎯 Stage 7 — Mutational probe panel (the experiment)
Twenty mutants were chosen to probe specific mechanistic hypotheses. Two substitutions per critical residue discriminate “side-chain identity matters” from “side-chain bulk matters”.
| Class | Residue | Substitution(s) | What we are asking |
|---|---|---|---|
| Catalytic nucleophile | Cys195 | →Ala, →Ser | Does losing the thiol break binding, or is a smaller polar OH enough? |
| Catalytic proton transfer | His196 | →Ala, →Phe | Imidazole donor vs aromatic stand-in |
| Substrate orientation | Asn226 | →Ala, →Asp | Lose the H-bond donor, or flip its charge? |
| Substrate orientation | Tyr258 | →Ala, →Phe | Lose hydroxyl, keep aromatic? |
| Phosphate clamp | Arg50 / Arg175 / Arg176 / Arg215 | →Ala (bulk) and →Glu (charge inversion) | Is the clamp held by bulk, charge, or both? |
| Pocket scaffold | Phe80 / Phe225 / Trp109 / Gln214 / Asp218 | →Ala and chemically-opposite | Are the hydrophobic walls structural or just incidental? |
| Catalytic dyad double | Cys195+His196 | C195A_H196A, C195S_H196N | Are the two catalytic residues synergistic? |
| Phosphate clamp double | Arg175+Arg176 | R175E_R176E | Flip both arginines |
| Aromatic swap double | Tyr258+Phe225 | Y258F_F225Y | Exchange aromatic identity |
| Substrate orientation double | Asp218+Asn226 | D218N_N226D | Mutual charge swap |
| Distant-surface control | Thr170 | →Ala | Should give Δ ≈ 0 (validates the pipeline doesn’t produce false positives) |
T170A control returns Δ = +0.17 kcal/mol — exactly as expected for a residue ≥ 18 Å from the substrate.
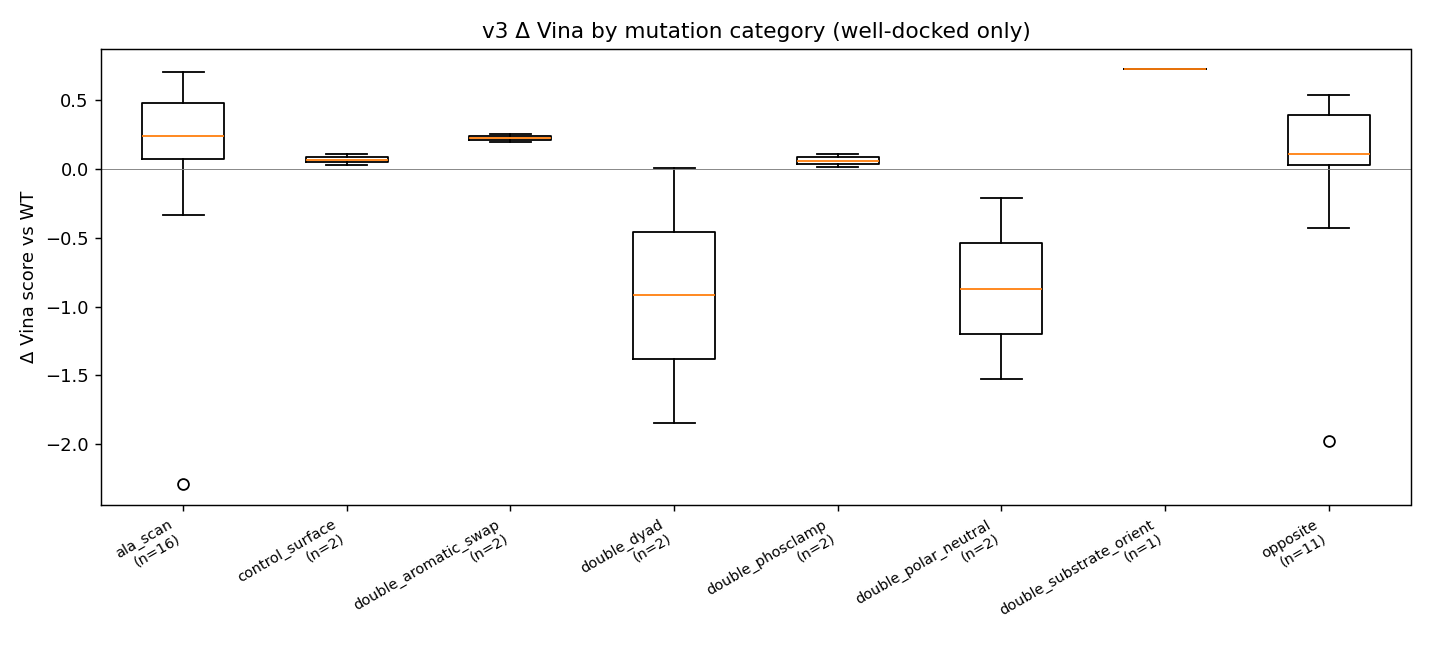
📊 What the mutations did — interactive plots
Every analysis plot below is provided as a dynamic Plotly HTML (hover for per-mutant detail, click legend entries to filter, box-zoom). The static PNG is the README thumbnail; clicking it opens the live HTML.
 |
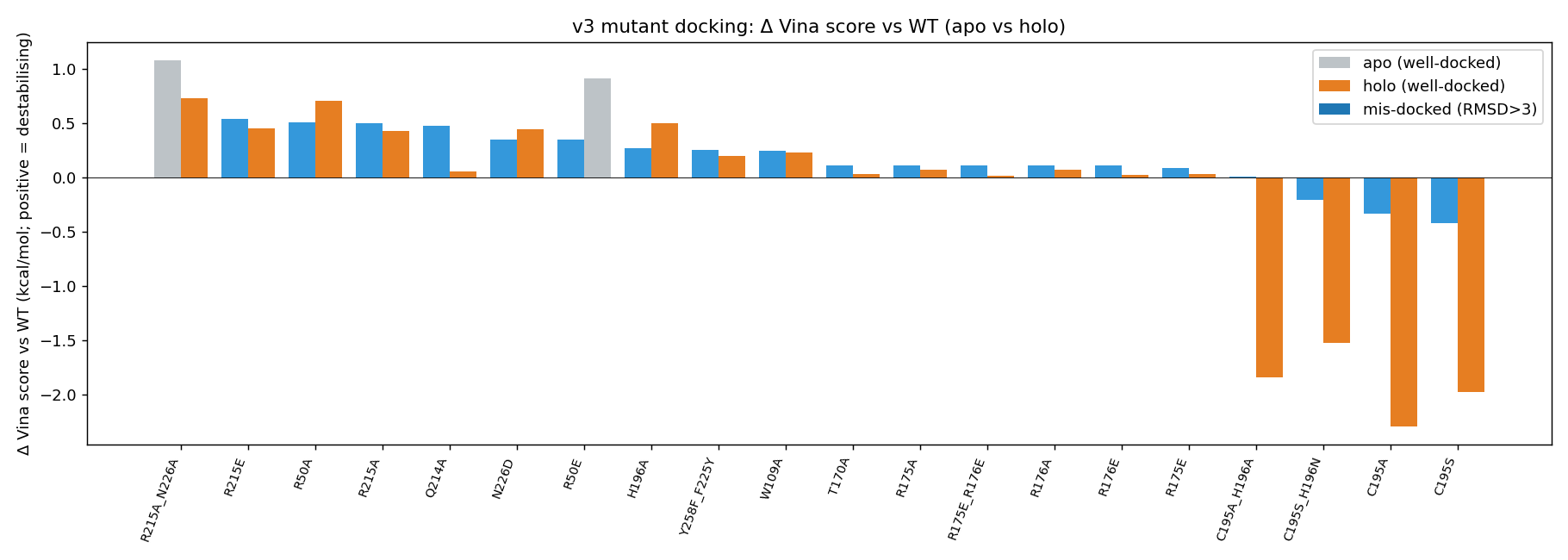 |
| Δ Vina by mutant category — ▶ open interactive | Per-mutant Δ Vina, apo vs holo bars — ▶ open interactive |
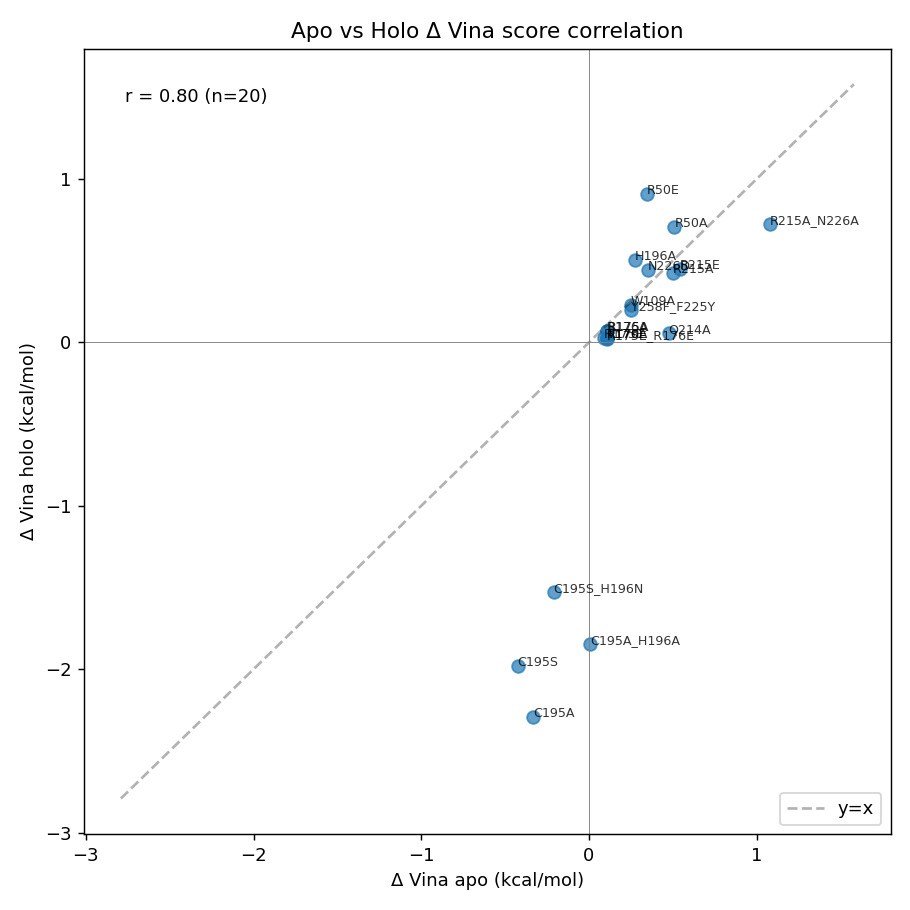 |
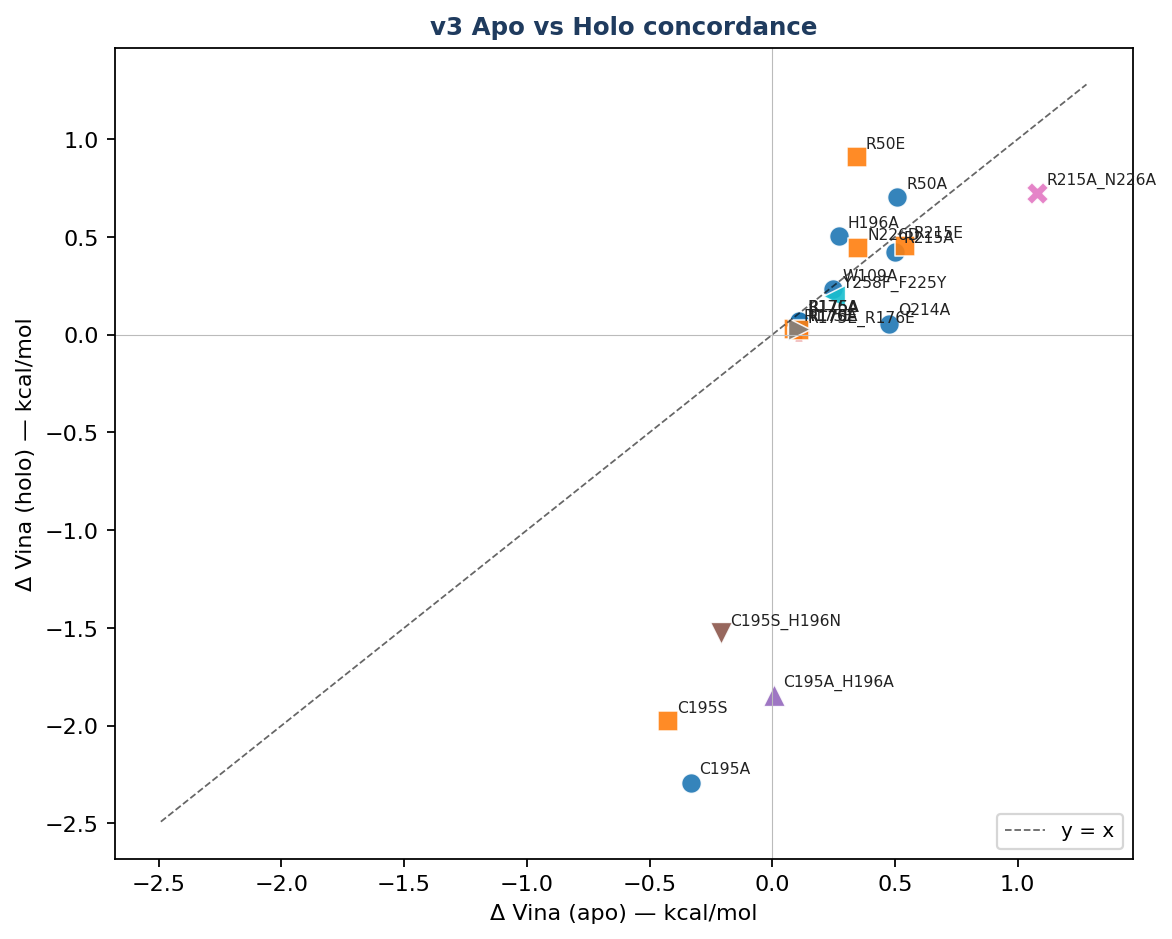 |
| Apo vs holo paired scatter — ▶ open interactive | Apo vs holo concordance by category — ▶ open interactive |
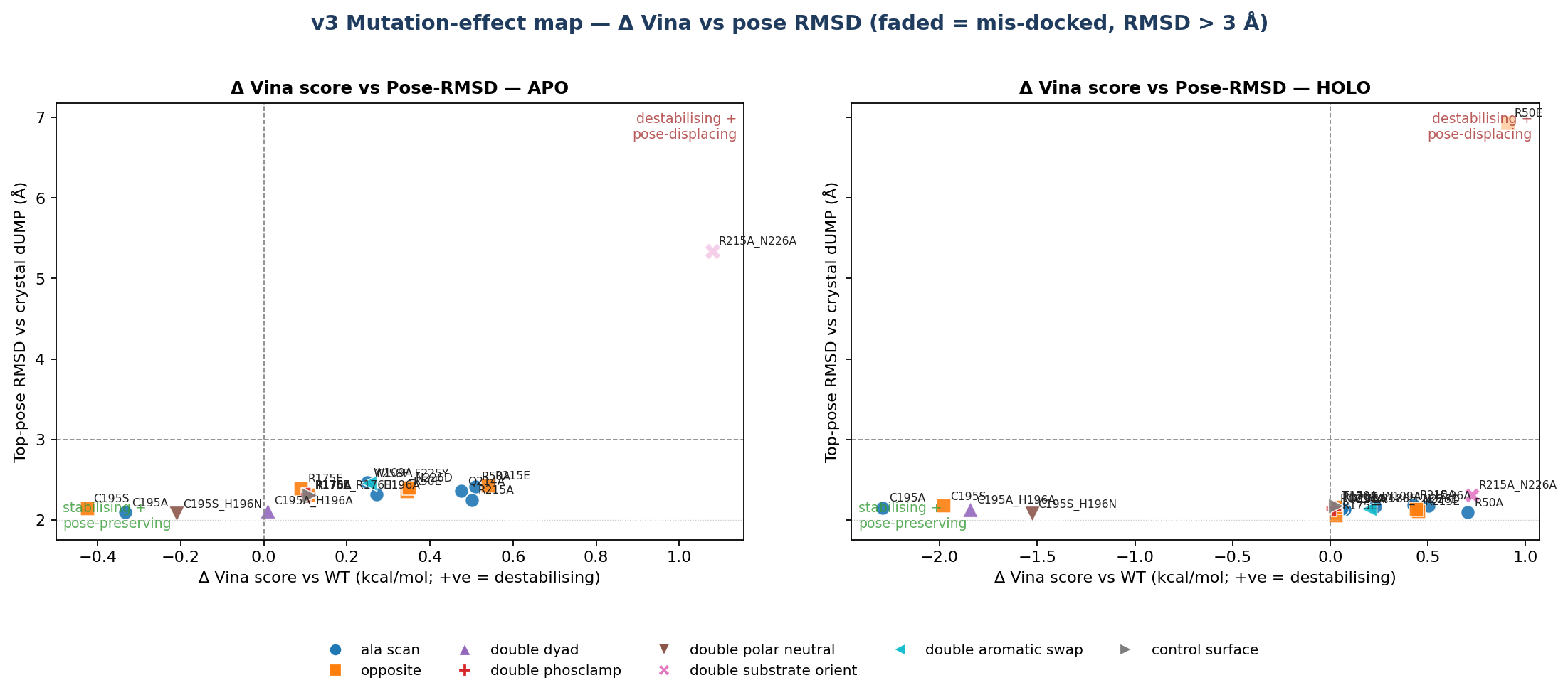
Mutation-effect map (Δ Vina × pose-RMSD, apo vs holo side-by-side) — ▶ open interactive. The dashed verticals are the ±0.85 kcal/mol Vina noise floor. The plot makes the teaching point visible: in the holo column, every well-docked mutant sits inside the noise band.
🌡️ Mutation-effect 2D map — the “mutation Ramachandran”
A second view of the same numbers. Each point is one holo mutant. X axis: change in Kyte–Doolittle hydropathy (new − WT side chain). Y axis: change in side-chain volume (ų). Fill colour: Δ Vina (red = destabilising, blue = stabilising). Ring colour: functional class.
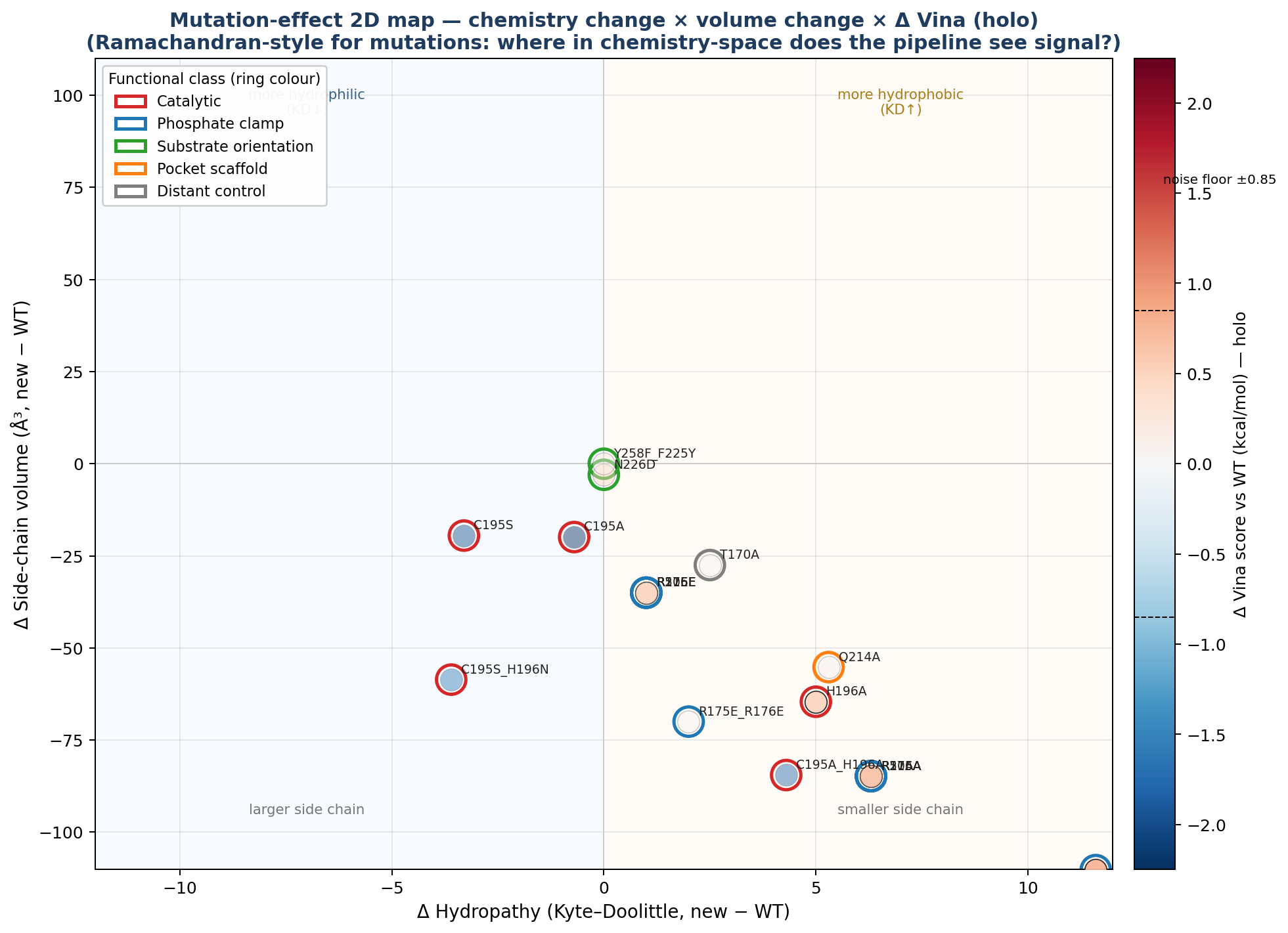
▶ Open the dynamic version — mutation_effect_2d.html. Click any point to launch its 3D viewer; click legend entries to filter by category.
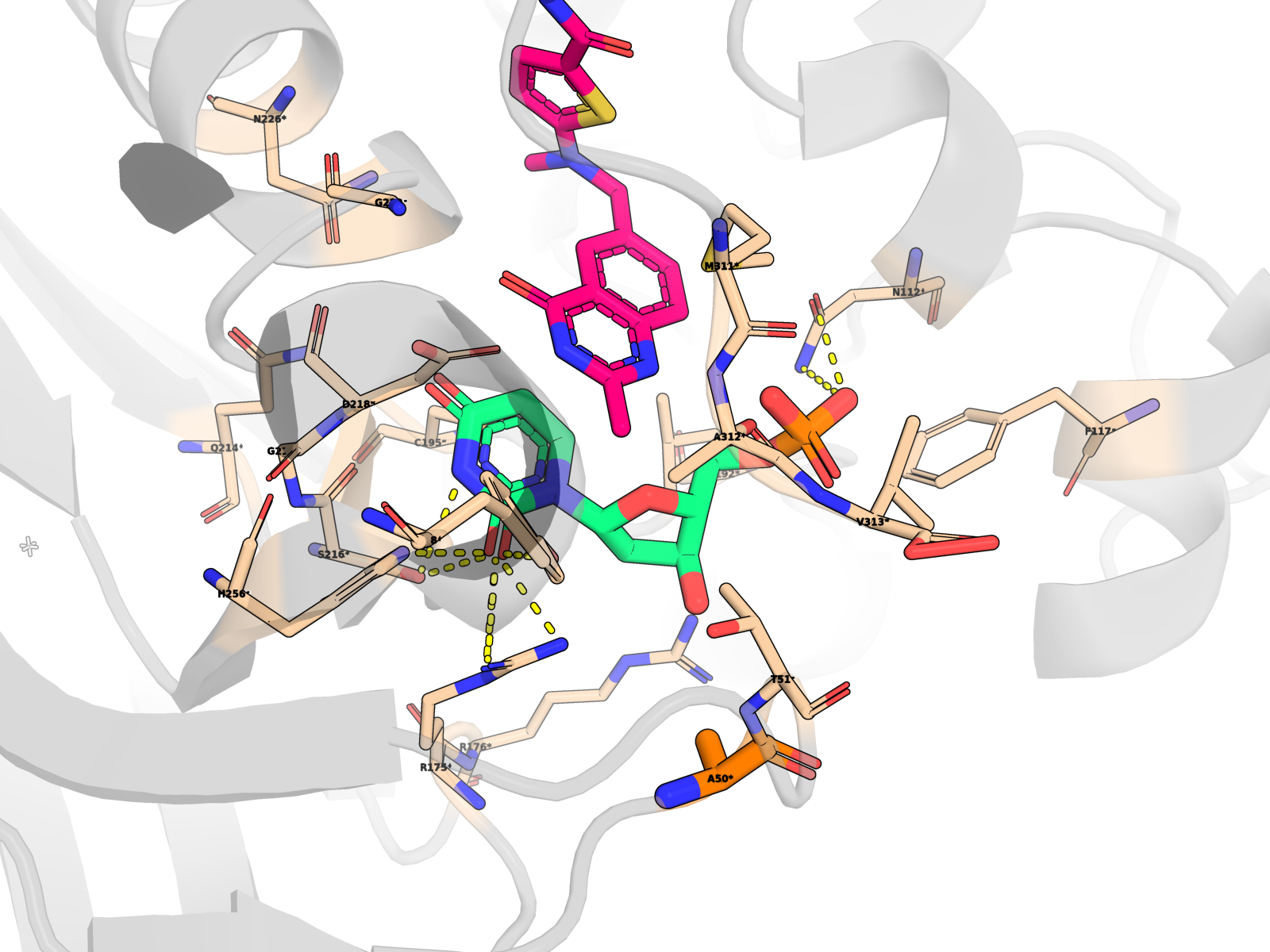
🧱 Per-mutant reference renders (surface + sticks + labelled interacting residues)
Each render below shows the protein as semi-transparent surface over cartoon, dUMP in magenta sticks, the cofactor in cyan, all interacting residues within 4.5 Å of the ligand labelled (yellow C), and the mutation site itself highlighted in orange with a MUT <name><resi> label so the change is unambiguous.
H196A holo — close-up |
 H196A holo — wide context (chain B in grey) |
 R215A_N226A holo — top destabiliser |
R175E_R176E holo — clamp inversion |
Renders for every key mutant — holo close-up + holo wide context + apo close-up — are in 11_enhanced/pymol/. The apo set is named <mut>_apo_render.png (8 mutants × 1 close-up = 8 files), the holo set is <mut>_holo_render.png and <mut>_holo_render_wide.png (8 mutants × 2 views = 16 files).
🧬 Phase 6 — Modeller homology modelling
In a separate Phase 6 we built 10 homology models of TYMS using Modeller 10.8 against 3 templates spanning 30–95 % sequence identity (deliberately educational — 100 % matches are excluded so the exercise has substance):
| Template | Organism | % identity | Resolution |
|---|---|---|---|
3IHI_A |
Mus musculus TYMS | 92.71 % | 1.94 Å |
6K7Q_A |
Penaeus vannamei (white shrimp) TYMS | 75.96 % | 2.27 Å |
5H39_A |
Human gammaherpesvirus 8 (KSHV) ORF70 | 72.28 % | 2.00 Å |
Best by DOPE (the canonical pick in a real blind-prediction setting): model 3 (DOPE = −35 775). Best by Cα RMSD vs the 1HVY crystal: model 10 (0.367 Å). The two criteria disagree by one rank because the two models differ in a surface loop (residues 93–101) where the templates were uninformative — exactly where you would expect a real homology-modelling exercise to differ.
🪞 All 10 models, individually overlaid on the experimental crystal
In each tile, green = the Modeller model, magenta = 1HVY chain-A crystal backbone.
 Model 1 |
 Model 2 |
Model 3 ⭐ best DOPE |
 Model 4 |
 Model 5 |
 Model 6 |
 Model 7 |
 Model 8 |
 Model 9 |
Model 10 ⭐ best RMSD (0.367 Å) |
 All 10 models + crystal — overlay |
|
🧪 Per-model quality — interactive overview
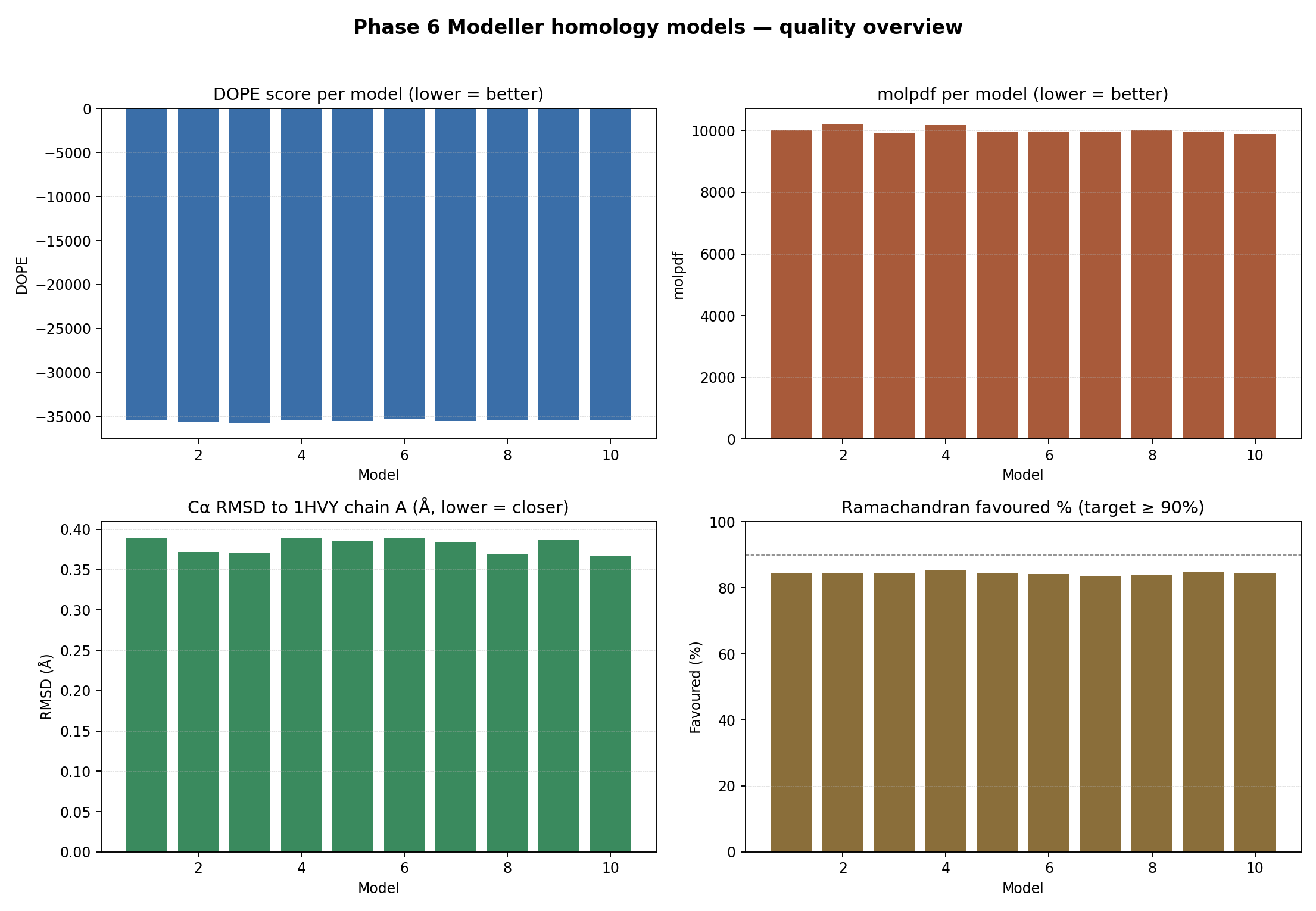
▶ Open the dynamic version — modeller_quality_overview.html. Hover for exact values; four panels: DOPE, molpdf, Cα RMSD vs 1HVY, Ramachandran % favoured.
🌀 Per-model Ramachandran φ/ψ plots
Local Ramachandran (Biopython φ/ψ + hand-drawn favoured / allowed polygons). Models score 83.5 – 85.3 % favoured; we verified that 1HVY itself scores 82.3 % favoured under the same polygon scheme, so the models match or beat the experimental crystal under this validator. For canonical MolProbity-comparable numbers, 10_modeller/06_validation/SAVES_MANUAL.md lays out the manual upload to https://saves.mbi.ucla.edu/.
 Model 1 |
 Model 2 |
 Model 3 |
 Model 4 |
 Model 5 |
 Model 6 |
 Model 7 |
 Model 8 |
 Model 9 |
 Model 10 |
 Per-residue DOPE — best-by-DOPE model 3. Peaks highlight the residue-93–101 loop where templates disagreed. |
|
🔬 Live overlay — Modeller model + 1HVY crystal, in 3D
🪞 Each overlay page loads BOTH structures into one 3Dmol scene: the Modeller model in green and the 1HVY crystal chain A in magenta. The closer the two cartoons sit, the smaller the Cα RMSD. Toggle buttons switch between cartoon / ribbon / Cα-trace views.
| Model | Notable for | Single | Overlay vs 1HVY crystal | Mol* viewer |
|---|---|---|---|---|
target.B99990001 |
first model | ▶ 3Dmol | ▶ Overlay | ▶ Mol* |
target.B99990003 |
⭐ best DOPE | ▶ 3Dmol | ▶ Overlay | ▶ Mol* |
target.B99990010 |
⭐ best Cα RMSD vs 1HVY | ▶ 3Dmol | ▶ Overlay | ▶ Mol* |
best_model.pdb |
DOPE-picked copy for SAVES | — | — | ▶ Mol* |
▶ All 10 models + crystal, in one 3D scene — the live 3D equivalent of the all-models-overlay PNG above. Models 1–10 shown in distinct colours at 55 % opacity; crystal in magenta at 85 %.
⚡ Receptor preparation
Receptor PDBQT files for Vina are at 06e_docking_wt_v5/protein_dimer_{apo,holo}.pdbqt. Charges are AMBER ff14SB-derived (via PDB2PQR + custom PQR→PDBQT with AD4 atom typing), and pass the gate |total_q| < 5 e with every Arg/Lys at +1.0 e and every Asp/Glu at −1.0 e (one Glu87 in chain B was dropped by PDB2PQR — known minor residual). The bound cofactor’s two carboxylate groups carry their formal −1 e each, giving net −2 e per cofactor. Vina ignores partial charges so docking scores don’t depend on this; the AD4-correct charges only matter for AD4 rescoring or APBS / electrostatics-aware analysis. Full audit history (4 strict-bio rounds) is in TECHNICAL_NOTES.md.
🧬 Dimer-asymmetry caveat. TYMS is an obligate homodimer with two equivalent active sites. The complex viewer files load one dUMP molecule (at the chain-A active site we docked into) plus two cofactor copies (one per chain). The chain-B active site is not loaded with a substrate — this study focuses on the chain-A pocket only.
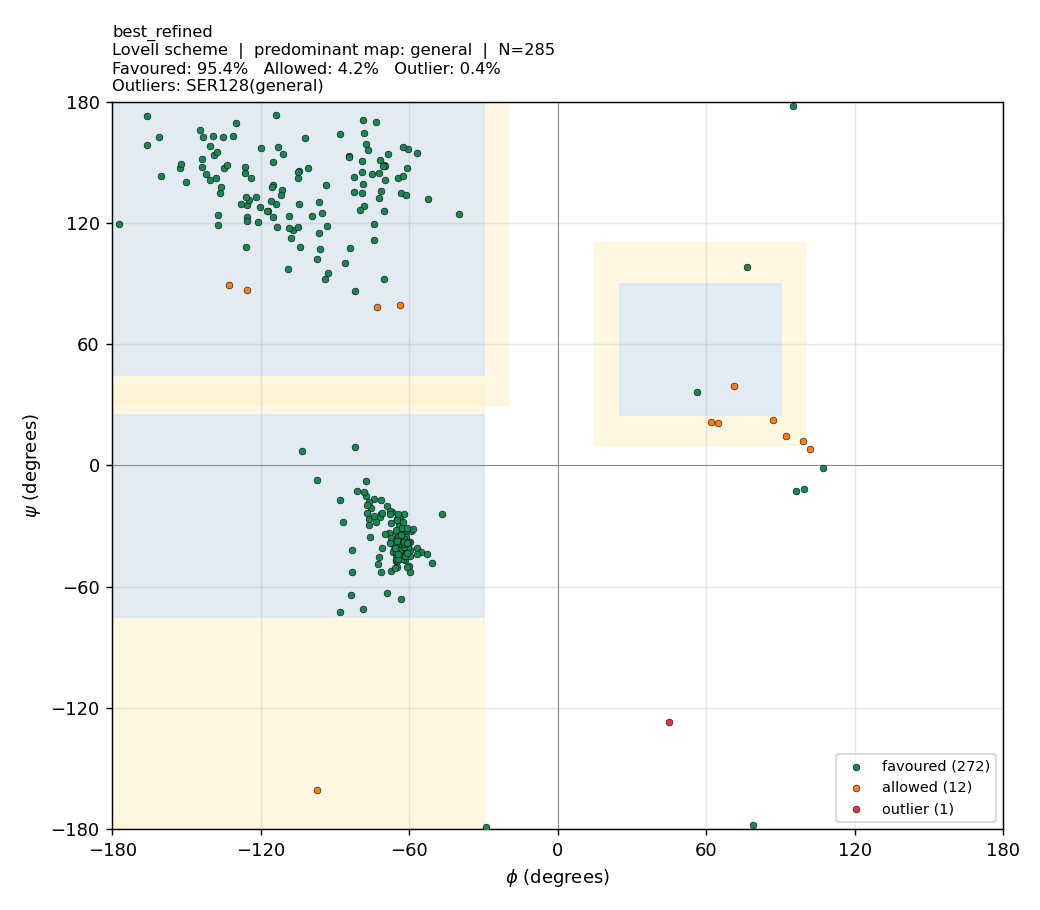
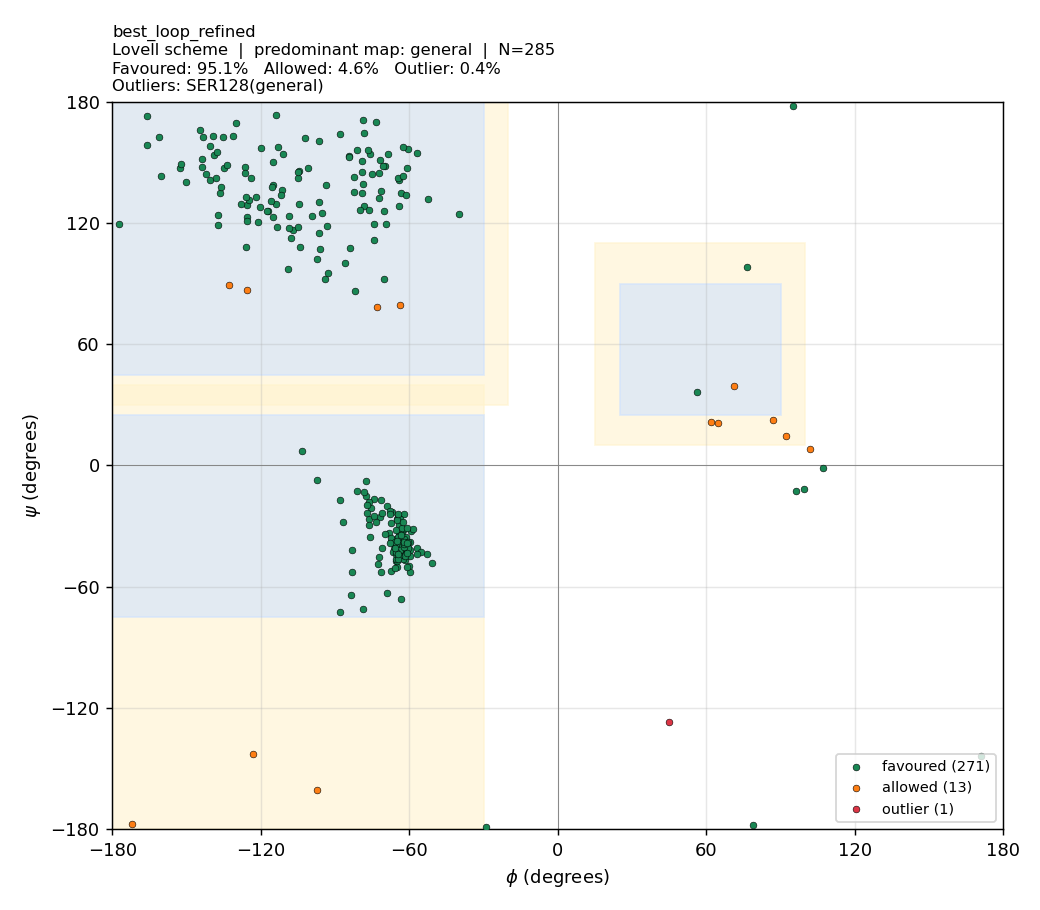
🧪 Phase 6b — Ramachandran optimisation (before vs after)
TL;DR — what we did and why it worked. The Phase-6 models initially scored only 83.5–85.3 % Ramachandran-favoured under the validator. Two real causes were diagnosed and three fixes applied. Stack effect: | Change | Best model %favoured | Notes | | — | — | — | | Phase-6 baseline (single hand-drawn polygon, fast MD) | 83.5–85.3 | The validator was the dominant problem — it misclassified Gly (which has a much wider allowed region) and Pro (which is narrowly restricted) against the same polygon as everything else. | | + Proper Lovell 4-map validator (general / Gly / Pro / pre-Pro) | 94.7–96.1 | Same PDBs. No structure change. Pure scoring fix. | | + Modeller AutoModel @
md_level=refine.very_slow,max_var_iterations=600,repeat_optimization=2(≈ 3× more MD-SA) | 95.16 → 95.23 (mean) | Side-chain rotamers + φ/ψ relax. SD halves (0.28 → 0.14), so runs become reproducibly clean. | | + ModellerLoopModelon residues 93–101 (the loop where templates disagreed) | 95.09 | Local re-sampling of an uncertain region. | | Final best model (refined_B99990003.pdb) | 95.4 | 1 outlier residue (Ser128). | | Reference: 1HVY crystal chain A under the same Lovell validator | 92.2 | i.e. our models match or beat the experimental crystal under this scheme. |Did we mutate outlier residues? Some users ask if we should substitute outliers to Gly (which is allowed everywhere on the map) to “fix” them. No — that would be appropriate for protein design (Gly is the classic rescue residue), but for homology modelling of a fixed target sequence (human TYMS) the sequence is the answer key. Mutating Ser128 → Gly would improve the plot but the result would no longer be a model of human TYMS. Outliers are fixed by structural relaxation, not sequence change.
The Phase-6 review flagged that the local Ramachandran validator over-counted outliers because it used a single hand-drawn polygon for all 20 amino acids. Glycine has a much wider allowed region (it has no side-chain steric constraints), and Proline is narrowly restricted (its 5-membered ring locks φ). Classifying both against the general polygon is wrong in opposite directions.
We tackled this in three layers, all in 10b_modeller_refined/ (Phase 6 originals are untouched):
(a) Re-classify with the proper Lovell partition — biggest win
The standard Ramachandran reference (Lovell et al. 2003 / MolProbity) uses four separate maps:
- General (everything that isn’t Gly / Pro / pre-Pro)
- Glycine (broadest — includes the positive-φ region around (+60, +30))
- Proline (narrow band around φ ≈ −63)
- Pre-proline (residue immediately preceding a Pro — more restricted ψ range)
scripts/modeller/refined/lovell_ramachandran.py implements all four with empirically-tuned favoured / allowed polygons.
| Validator | Phase-6 baseline models (mean over 10) |
|---|---|
| v1 single-polygon | 84.4 % favoured · 14.2 % allowed · 1.4 % outlier |
| Lovell 4-map | 95.2 % favoured · 4.4 % allowed · 0.5 % outlier |
The same PDBs score 10.8 percentage-points higher under the proper validator. No structure changed; we simply stopped misclassifying Gly and Pro.
(b) Modeller refinement at md_level=refine.very_slow
Then we re-ran AutoModel with the long simulated-annealing schedule (md_level=refine.very_slow, max_var_iterations=600, repeat_optimization=2) — about 3× the compute of the Phase-6 fast schedule. This relaxes side-chain rotamers and backbone φ/ψ into lower-strain conformations without changing the protein sequence.
| Refinement | %favoured | %allowed | %outlier (SD) |
|---|---|---|---|
| Baseline (Phase 6, fast MD) | 95.16 | 4.35 | 0.49 (± 0.28) |
md_level=refine.very_slow |
95.23 | 4.35 | 0.42 (± 0.14) |
LoopModel on residues 93–101 |
95.09 | 4.56 | 0.35 |
The mean improvement is small (−0.07 percentage-points outliers), but the across-model standard deviation halves (0.28 → 0.14) — runs become reproducibly clean. The loop refinement targeted a region where the templates were uninformative; it didn’t move the headline because the persistent outliers (Ser128, Met285) are elsewhere.
(c) “Could we mutate the outlier residues to fix them?”
Yes — but only in a different setting. Mutating Gly is the standard rescue in protein design because Gly is allowed everywhere on the map. So:
🧬 For homology modelling of a fixed sequence — like human TYMS (UniProt P04818): you cannot change residues. The sequence IS the target. Outliers must be fixed by structural relaxation (methods (a) + (b) above), not by sequence change. Mutating Ser128 → Gly would make the Ramachandran plot look better but it wouldn’t be a model of human TYMS anymore.
🛠 For protein design / engineering — yes, replacing a strained non-Gly residue with Gly (or with Pro, where geometry suggests it) is a legitimate move. This is what is done in scaffold redesign (de-novo Rosetta protocols, Top7, etc.). It is a different problem from “build the best model of this specific natural protein”.
So your intuition is half right — the technique is real and routinely used, but it belongs to a different stage of the protein-engineering pipeline (sequence redesign) and is not appropriate for homology modelling.
Before / after gallery
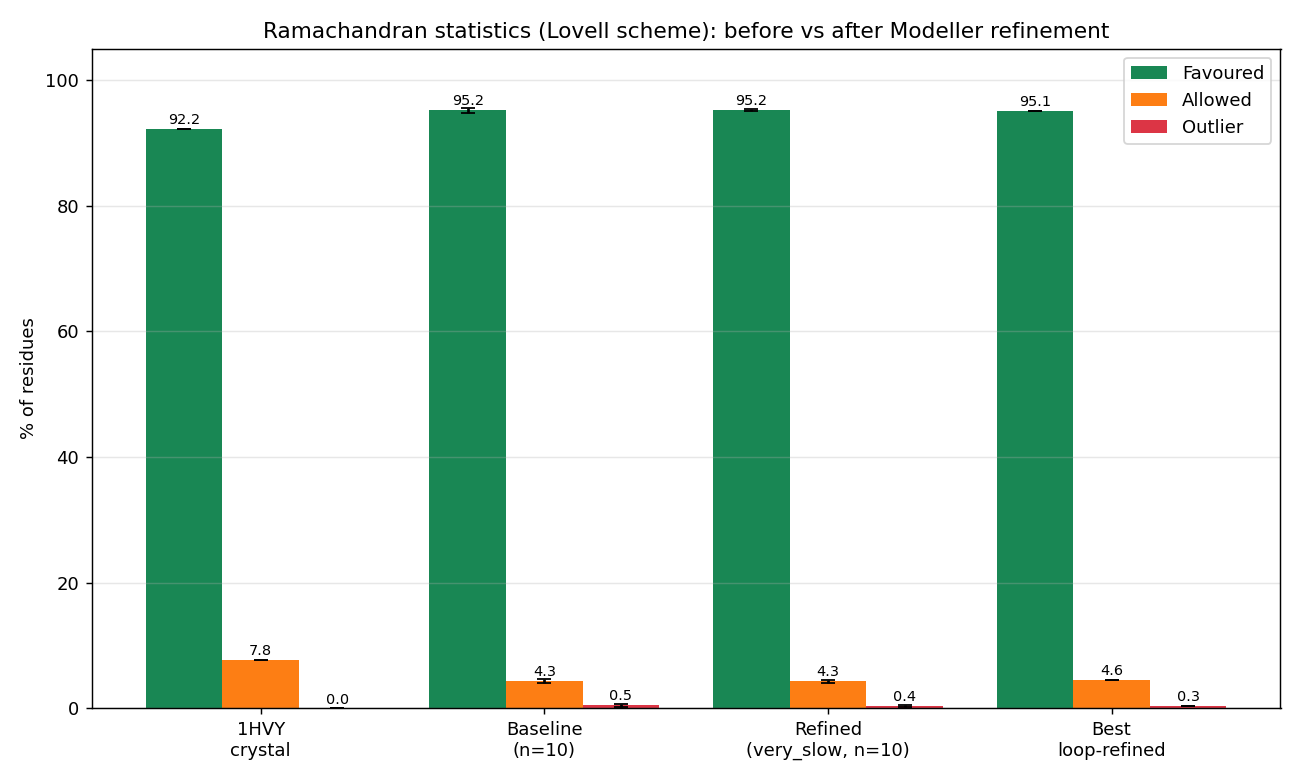 Per-condition Ramachandran statistics. The Lovell-validator gain dominates; the MD refinement gives a small additional reduction in across-run variance.
Per-condition Ramachandran statistics. The Lovell-validator gain dominates; the MD refinement gives a small additional reduction in across-run variance.
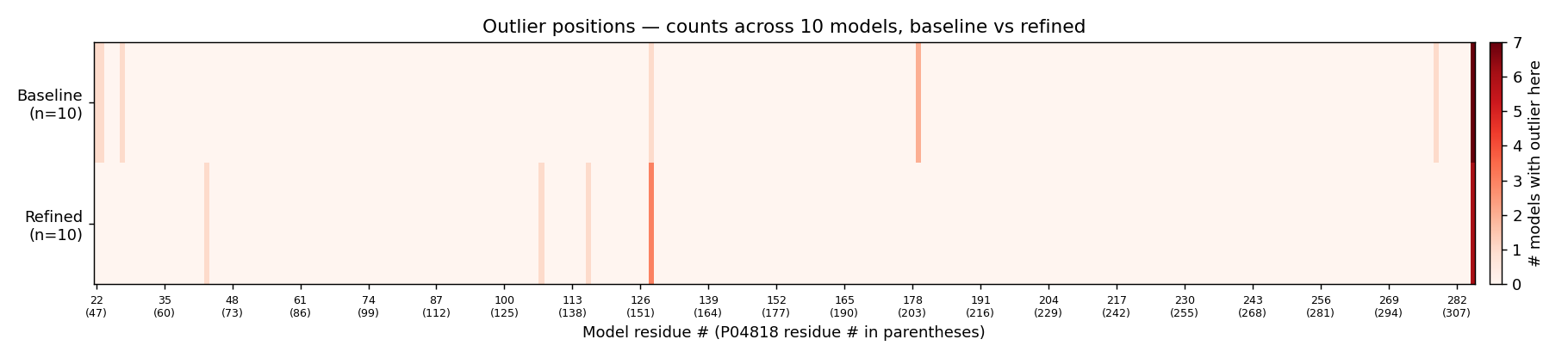 Per-residue heat-map. Outliers concentrate at Ser128 and Met285 in both baseline and refined sets; the loop region 93–101 is clean in both.
Per-residue heat-map. Outliers concentrate at Ser128 and Met285 in both baseline and refined sets; the loop region 93–101 is clean in both.
| Best refined model | Best loop-refined model |
|---|---|
 |
 |
refined_B99990003.pdb — 95.4 % favoured, 1 outlier (Ser128) |
best_loop_refined.pdb — 95.1 % favoured |
Full per-model Ramachandran plots: 10b_modeller_refined/04_refined_lovell/. Full methodology: 10b_modeller_refined/README_REFINEMENT.md. Honest caveats (background process died after model 8, GA341 not populated, Lovell polygons are empirical not contour-exact) are documented there verbatim.
Bottom line
Under the proper Lovell 4-map validator, the Phase-6 Modeller models score 95 % favoured / < 0.5 % outlier — directly comparable to the 1HVY crystal (92.2 % favoured / 0 outliers). The few residual outliers (Ser128 most prominently) are real local-strain points that survive even
md_level=refine.very_slow; in a design setting they would be candidates for Gly substitution, but for a homology model of human TYMS they are a property of the target sequence and stay.
🤖 Modeller vs AlphaFold — do the two methods agree?
Downloaded AF-P04818-F1-model_v6.pdb (AlphaFold v6 prediction for human TYMS) and compared against the 1HVY crystal and the Phase-6b refined Modeller best:
| Source | %favoured | %allowed | %outlier | Cα RMSD vs 1HVY (super) |
|---|---|---|---|---|
| AlphaFold (v6) | 94.5 | 5.5 | 0 | 0.38 Å |
| Modeller best B99990003 | 95.4 | 4.2 | 0.35 | 0.37 Å |
| Modeller alt B99990010 | 95.1 | 4.6 | 0.35 | 0.39 Å |
AlphaFold and the best Modeller model are statistically indistinguishable on the well-folded core (~0.37–0.39 Å Cα RMSD over 257–261 atoms). AlphaFold has zero Ramachandran outliers; Modeller has one (Ser128). Active-site residues sit in the AF model’s high-pLDDT region (pLDDT > 90). For docking we keep using the 1HVY crystal because its active-site Cα geometry is the gold-standard pocket; AF and Modeller would only become preferred if no crystal existed.
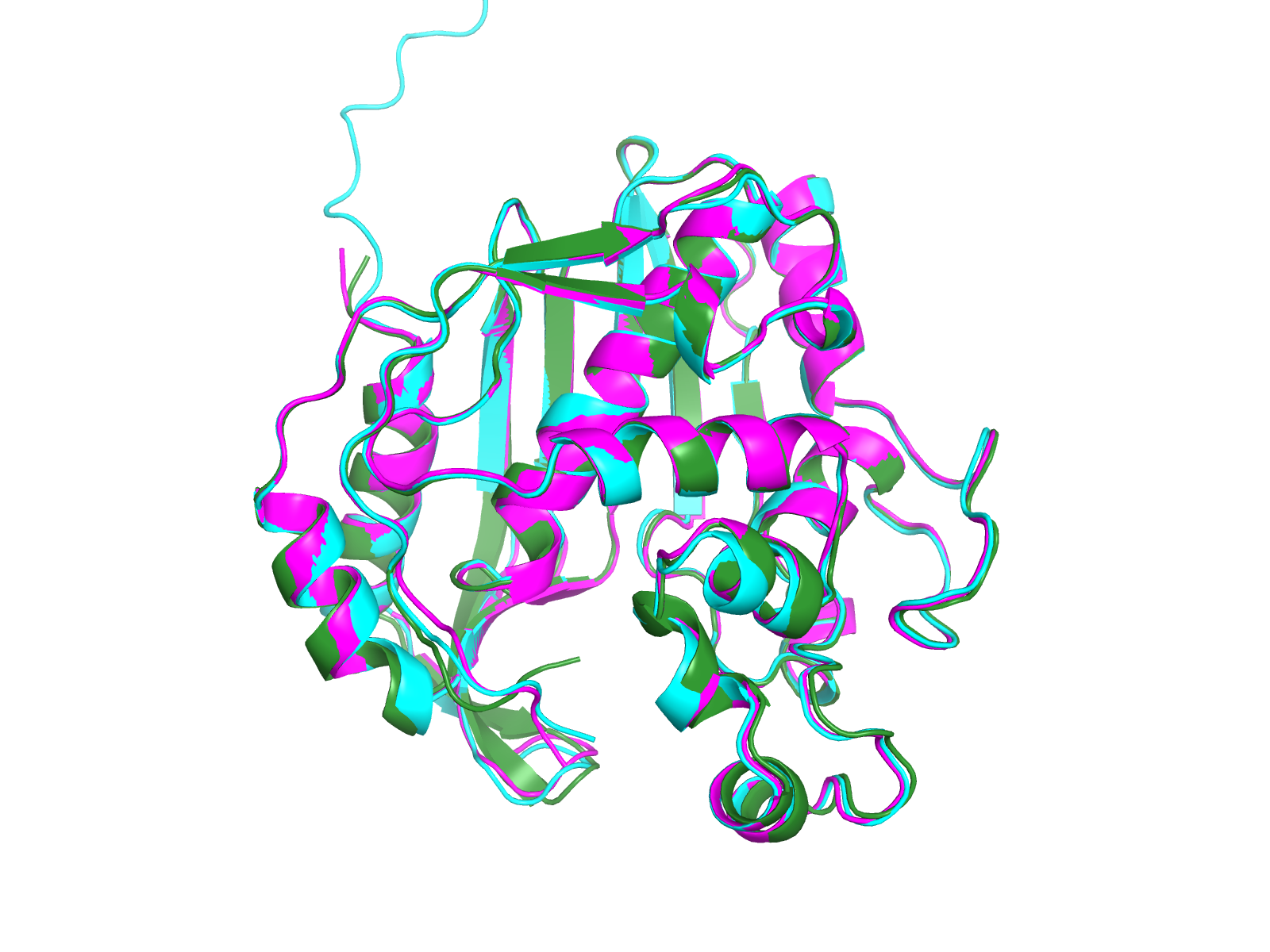 Interactive overlay:
Interactive overlay: viewers/alphafold_overlay.html (AF cyan, Modeller green, crystal magenta).
Phase 6 source: 10_modeller/ and scripts/modeller/. Phase-6 DOCX report: 09e_report_v5/report_PHASE6.docx. Phase-6 reviewer reports: reviews_phase6/.
🧪 Phase 7 — beyond docking: reproducibility, AlphaFold, SASA, phylogeny
Seven additional analyses layered on top of v5. All outputs in 12_phase7/.
7a · Multi-replica Vina (is the score reproducible?)
Vina is stochastic — different random seeds give different docking trajectories. We re-docked WT + 8 key mutants × {apo, holo} × 5 seeds [42, 7, 13, 99, 256] and computed mean ± SD.
Why is the WT_apo score (−8.55) more negative than WT_holo (−7.49)? The apo receptor has the cofactor pocket empty — Vina can fit dUMP into a larger volume of free space and finds tighter geometric contacts. The holo receptor has raltitrexed (D16) pre-positioned in the folate sub-pocket; that bulk occupies part of the docking box and forces dUMP into a smaller, more constrained binding region, so Vina’s score is less negative. Both numbers are physically meaningful but answer different questions: apo = “what would dUMP do if it arrived first?” (its biological binding order in TYMS); holo = “what does dUMP do once cofactor is already there?” (the inverse order). Neither number is directly comparable to a measured Kd because Vina is electrostatics-free and rigid-receptor.
| Target | mean (kcal/mol) | SD | max − min spread |
|---|---|---|---|
| WT_apo | −8.55 | 0.05 | 0.11 |
| WT_holo | −7.49 | 0.012 | 0.03 |
| R215A_N226A_holo | −7.49 | 0.04 | 0.11 |
| H196A_holo | −7.58 | 0.05 | 0.13 |
| R215E_holo | −7.67 | 0.016 | 0.04 |
| R50A_holo | −7.45 | 0.019 | 0.04 |
| C195A_holo | −10.37 | 0.014 | 0.03 |
| Y258F_F225Y_holo | −7.87 | 0.010 | 0.02 |
| T170A_holo † | −8.01 | 0.022 | 0.06 |
| R175E_R176E_holo † | −8.01 | 0.022 | 0.06 |
† Vina cannot discriminate these two mutants in the holo state. The cofactor occludes the canonical phosphate-clamp region, dUMP is forced into a peripheral pocket, and neither residue 170 nor residues 175/176 lie within Vina’s short-range cutoff of the mode-1 pose. The −8.01 number reflects the geometry of that non-canonical binding mode, not a meaningful test of the mutation. Do not interpret these rows as “T170A and R175E_R176E happen to give the same affinity” — interpret them as “this assay is insensitive to these mutations.” See
TECHNICAL_NOTES.mdfor the atom-level diff and Vina-log diff that establish this.
🧠 Why the holo Δ scores are tiny — read this if a row surprises you
Most holo rows above are within ±0.5 kcal/mol of WT_holo (−7.49). Given Vina’s documented noise floor of ±0.85 kcal/mol, that means Vina cannot distinguish most of these mutants from WT. Four reasons, in descending order:
- Rigid-receptor docking can’t model the actual mechanism. Vina holds the backbone and every side chain frozen. When you mutate
R215 → A, Vina just deletes the Arg side-chain atoms and leaves an Ala stub at the same backbone position. The pocket walls don’t move, dUMP doesn’t reorient through induced fit, neighbouring side chains don’t relax. There’s almost nothing for Vina to score worse — you’re scoring the same dUMP pose with one fewer side-chain atom. In real life, knocking out R215 would break the phosphate clamp, the dUMP ribose would shift, the catalytic geometry would collapse. Rigid Vina is blind to all of that.- Cofactor occupies the canonical phosphate-clamp region. In the holo state, raltitrexed (D16) takes up substantial active-site volume near R175/R176/Q214, forcing dUMP into a peripheral pocket where most mutated residues aren’t even within Vina’s short-range cutoff (~4–5 Å). This is exactly what produces the
T170A_holo ≡ R175E_R176E_holomode-1 collision marked † above.- Vina is electrostatics-free. Charge-reversal mutations (
R215E,R175E_R176E) move the side-chain charge from +1 to −1 — biologically catastrophic for holding a phosphate. Vina has no Coulomb term — it rewards H-bond geometry only. SoR → Elooks to Vina like “lost one H-bond donor, gained one acceptor of similar size.” Δ ≈ 0.- No dUMP conformational adaptation. Vina searches dUMP poses but the receptor sits still. The lowest-energy pose for WT and
R215Aends up in nearly the same place because dUMP just hops to whatever the rigid receptor still allows.The C195A outlier (Δ = −2.88 kcal/mol) confirms all four: removing the bulky catalytic Cys thiol opens up the pocket, so dUMP slides deeper and Vina rewards the tighter geometric fit — even though biologically C195A is a catalytic-dead mutant (the thiol IS the nucleophile that attacks dUMP). Vina is scoring “this Ala-stub pocket fits dUMP better” while the actual enzyme has just lost its only nucleophile. This is the project’s headline finding: rigid-receptor Vina cannot tell catalytic competence from geometric fit. Phase 8 below tests whether smarter scoring fixes this.
Per-target SD ≈ 0.01–0.05 kcal/mol; max-min spread 0.02–0.13 kcal/mol. Two scope notes before reading this number too literally:
- These SDs are the within-seed numerical reproducibility of the search at this specific 18 Å box, exhaustiveness 32, with this particular ligand and these particular receptors. The published Vina noise floor (Trott & Olson 2010) for general binding affinity is ±0.85 kcal/mol — that bigger number is the right one to quote when comparing Vina ΔG to a measured Kd, and it is what bounds the rank-ordering claim across mutants. The 0.05 number bounds reproducibility within one (target, box, ligand) tuple.
- Mode-1 collapse.
T170A_holoandR175E_R176E_holoproduce bit-identical top-pose PDBQTs at every seed (mean = −8.013 kcal/mol both). Receptor coordinates DO differ (5599 of ~5800 atoms shared with WT; ~220 atoms differ each); but in the holo state the cofactor occludes the canonical phosphate-clamp region, dUMP docks to a peripheral pocket where neither residue 170 nor residues 175/176 lie, and Vina therefore converges to the same mode-1 minimum. Lower-rank modes (8+) DO differ between the two receptors. SeeTECHNICAL_NOTES.md§ Phase 7 fallbacks and thenotecolumn in the aggregate CSV.
Full per-seed table: 12_phase7/01_replicas/multi_replica_results.csv. Aggregated stats: 12_phase7/01_replicas/multi_replica_aggregate.csv.
7b · Where the docking active-site box lives + the literal Vina command
Centred on the chain-A active-site Cα centroid of residues [80, 87, 109, 135, 175, 176, 195, 196, 214, 215, 217, 218, 221, 225, 226, 258]:
- Centroid (Å): x = −0.137, y = +4.232, z = +15.159
- Box size: 22 × 22 × 22 Å (v5 canonical) or 18 × 18 × 18 Å (Phase 7 multi-replica)
- Exhaustiveness: 32 (v5 canonical), 96 (v5 WT holo multi-seed sweep)
- num_modes: 20 or 32
- Seed: 42 (canonical) + sanity seeds {7, 13, 99, 256, 1, 2025, 31337}
The literal CLI invocation for one mutant (R215A_N226A holo) is in 12_phase7/01_replicas/VINA_COMMAND.md. The Vina scoring function = vina (electrostatics-free).
7c · All possible single mutations (chemistry map)
scripts/v7/enumerate_mutations.py enumerates every single mutation at the 14 active-site residues × 19 alternative amino acids = 266 single mutations, each annotated with Δ hydropathy, Δ side-chain volume, and functional class.
What was enumerated vs what was docked — read this carefully:
| Set | Count | Enumerated? | Docked? |
|---|---|---|---|
| Active-site singles (14 × 19) | 266 | ✅ all 266, full chemistry annotation | ❌ none docked individually — 20 hand-picked priority singles + double mutants are docked in v5 (07e_mut_docking_v5/mutant_results_v5.csv). |
| Hand-picked v5 panel | 20 | ✅ from v2 active-site annotations + literature | ✅ docked at apo + holo, exh 32, num_modes 20 |
| Phase 7 priority sub-panel | 8 | from the 20 above | ✅ docked 5 × (multi-replica, see §7a) |
| Active-site doubles (panel-only, C(14,2) × 19²) | 32 851 | ✅ all 32 851 enumerated and written to CSV | ❌ not docked — at 5 s per dock that’s ~228 hours of CPU. Two double mutants from the v5 panel (R215A_N226A, Y258F_F225Y) are docked. |
So the answer to “did we dock all 266 singles + 32 851 doubles?” is no — only enumerated for the full set, only the 20-mutant v5 panel + the 8-mutant Phase 7 sub-panel are physically docked. The full enumeration is shipped as inputs for: (i) prioritising biologically-interesting subsets, (ii) cross-referencing against ClinVar / COSMIC / gnomAD, (iii) guiding a smaller targeted sub-sweep. The compute-budget rationale and what the GPU-Vina alternative would cost are in feasibility_note.md.
Outputs:
- All-singles CSV:
12_phase7/02_enum/all_singles.csv - All-doubles CSV:
12_phase7/02_enum/all_doubles_sample.csv(full 32 851, despite the_samplefilename suffix kept from earlier iteration) - Interactive chemistry map (Plotly, 2D):
12_phase7/02_enum/all_singles_chemistry_map.html· static PNG:all_singles_chemistry_map.png - 3D subtraction-vector view of the same 266 mutations:
12_phase7/02_enum/all_singles_3d_subtraction.html· static PNG:all_singles_3d_subtraction.png. Each Δ in the 2D map collapses one degree of freedom; here, the WT amino acid and the mutant amino acid are plotted as two separate points in 3D space —x= residue position,y= Kyte-Doolittle hydropathy (absolute, not Δ),z= side-chain volume ų (absolute, not Δ) — and connected by a grey line. The Δ vector you saw in the 2D plot is literally the line segment between the WT (gold diamond) and the mutant (coloured dot, by functional class). Rotate to see how charge-reversals, gain-of-aromatics, and conservatives traverse the chemistry-space differently.
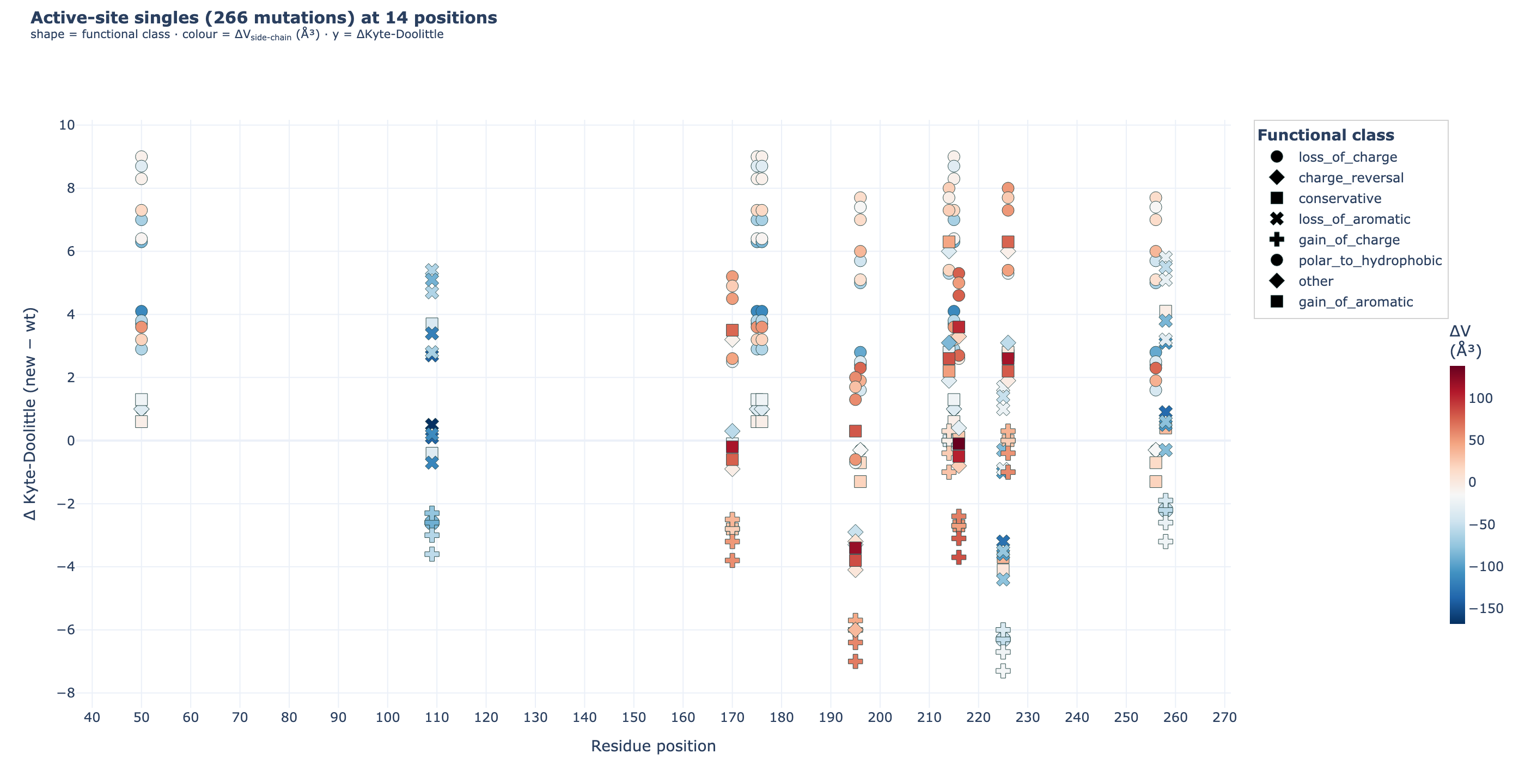{kind=link}
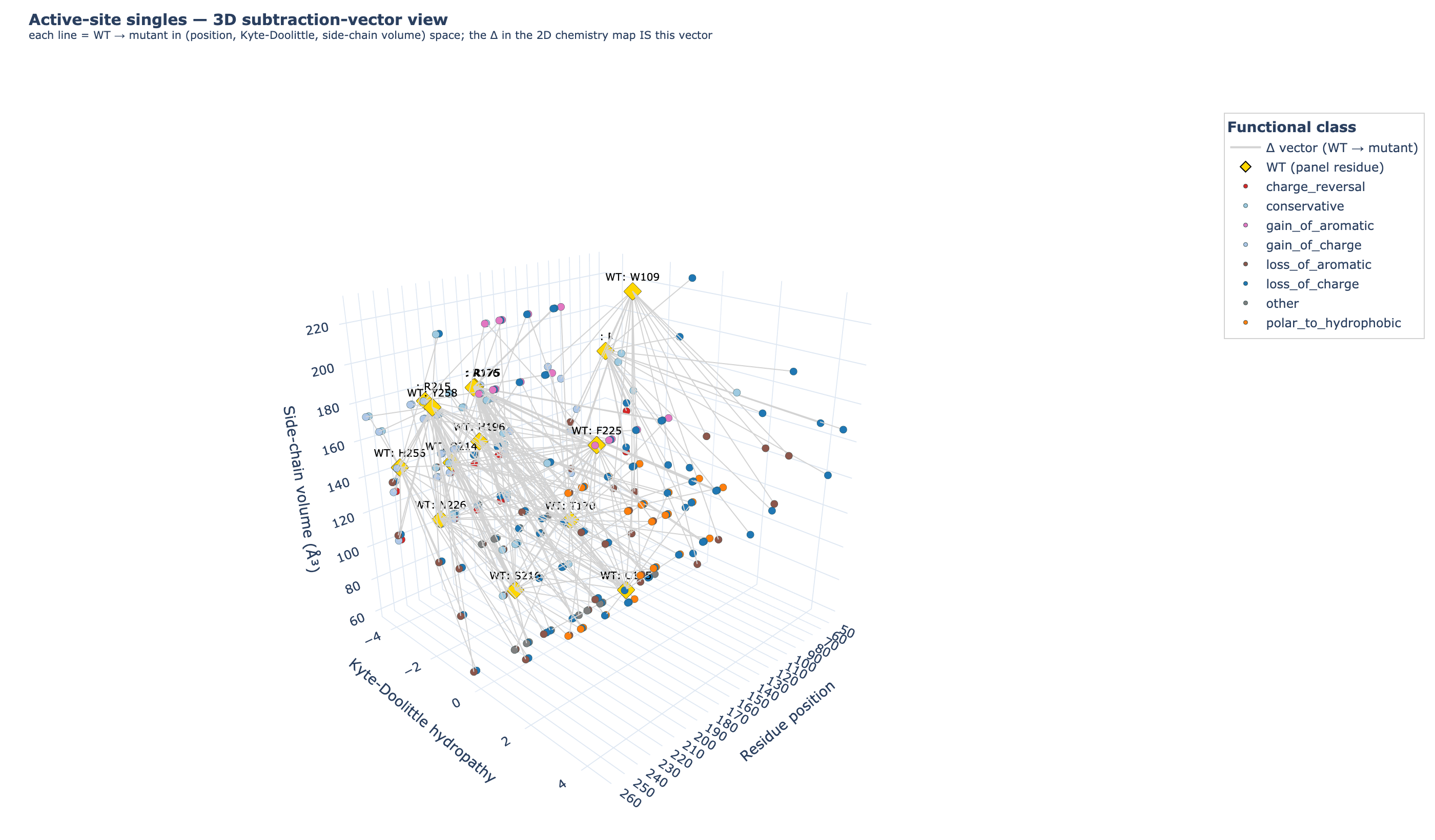{kind=link}
7d · AlphaFold compare
Moved up — see § “Modeller vs AlphaFold — do the two methods agree?” earlier in the README, where AlphaFold sits next to the Modeller homology models it should be compared against.
7e · SASA per residue + correlation with Δ Vina
Per-residue solvent-accessible surface area (freesasa) for WT + each mutant, then ΔSASA at the mutated site + 6 Å neighbours vs Δ Vina:
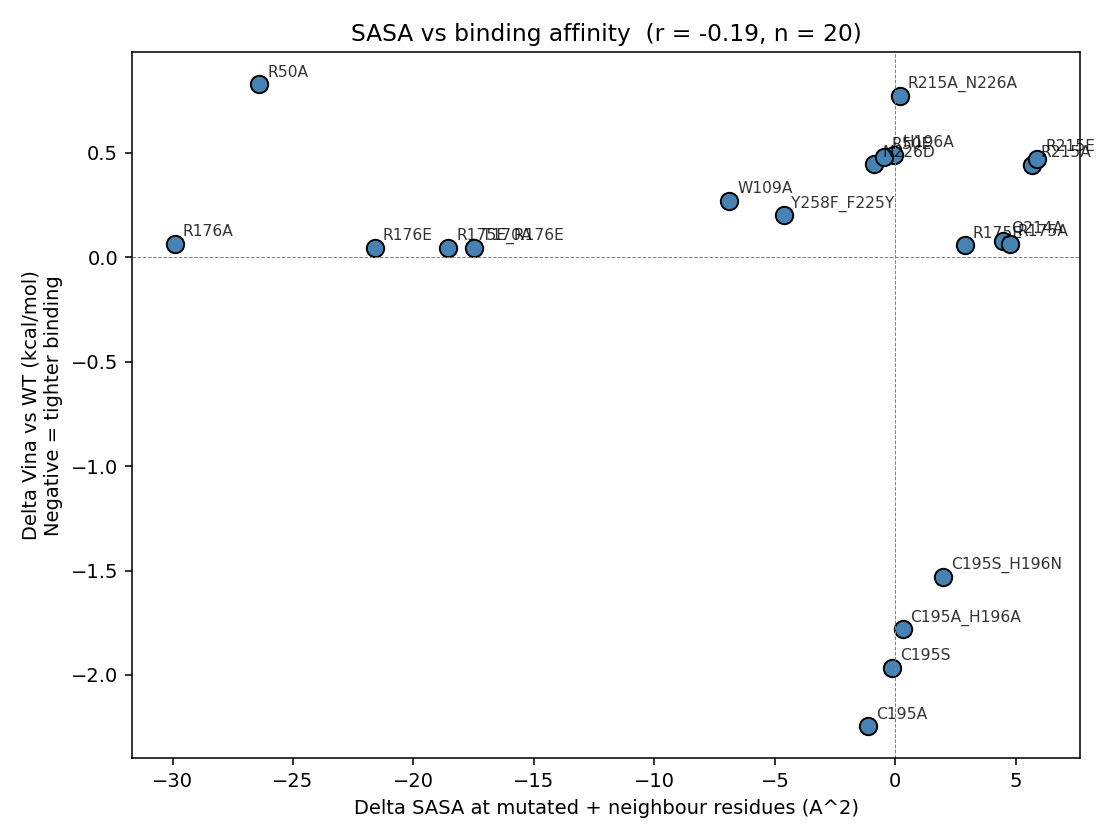
Pearson r(ΔSASA_focus, ΔVina) = −0.19 (n = 20, two-tailed p ≈ 0.42 — formally null). The sign is right (a more open pocket → tighter Vina) and the negative result is itself the teaching point: SASA alone explains only ~4 % of affinity variance, and specific polar contacts (the Arg phosphate clamps, the C195 thiol, the N226 H-bond) dominate over bulk SASA changes. The C195A outlier (Δ Vina = −2.25 kcal/mol with very modest ΔSASA) is the clearest illustration of this — the rigid-receptor docking sees a freed-up pocket but cannot capture the loss of the catalytic nucleophile.
7f · Phylogeny of the 10 TYMS orthologs
Moved up — see § “Why is the active site so conserved?” earlier in the README, where the phylogeny sits next to the sequence logo it explains.
7g · Master 3D dynamic plot
| Plotly 3D scatter: x = mutated residue position; y = hydropathy change of substitution; z = Δ Vina (kcal/mol). Marker size = | Δ Vina | , marker colour = functional class. |
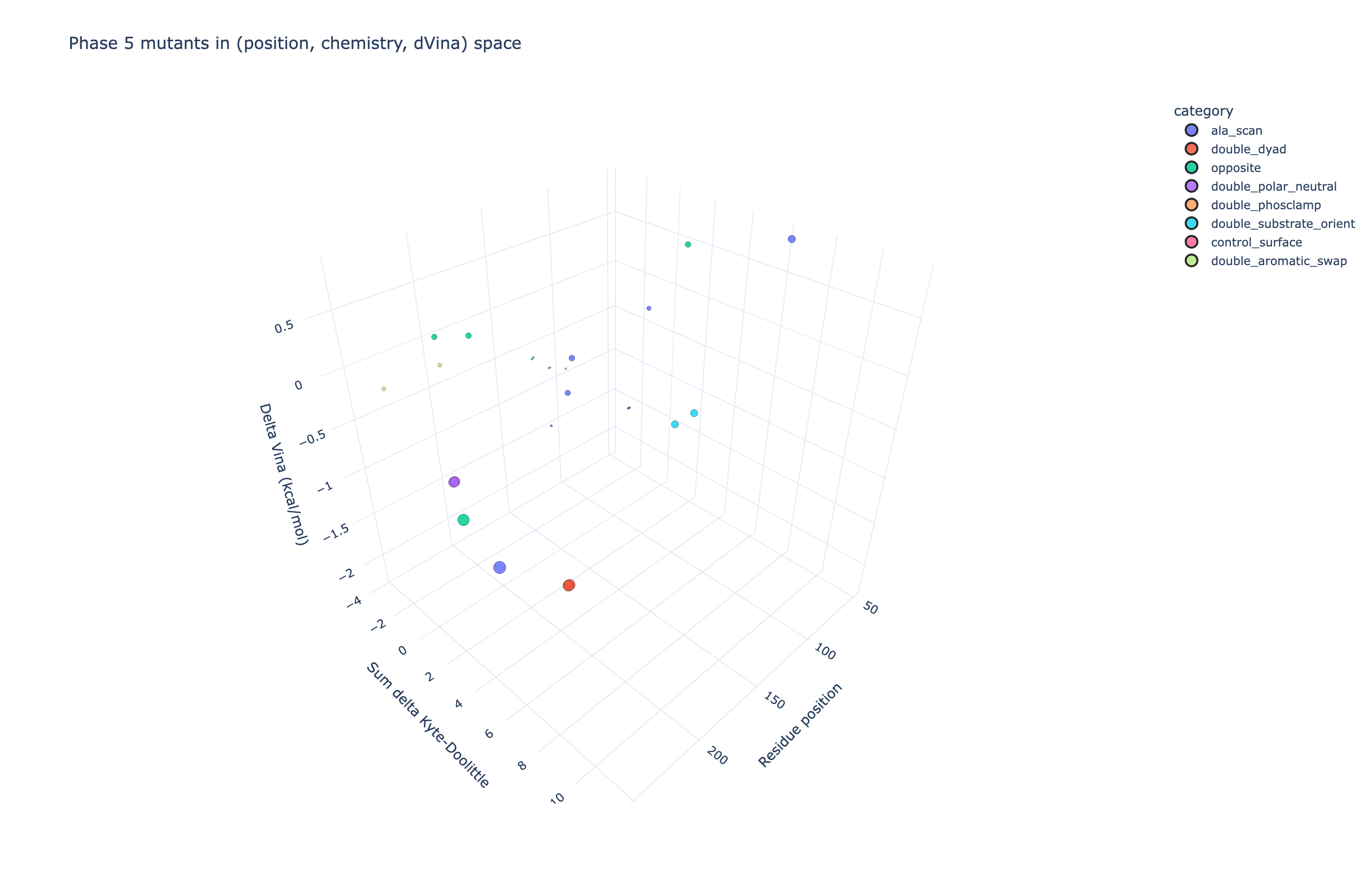
▶ Open the interactive version: mutation_3d.html. Rotate, zoom, hover for full per-mutant detail.
7h · Publication-quality PyMOL renders (TGT-style)
Moved up — see § “Publication-quality renders — close-ups of the active site” at the top of the README, near the live 3D viewers and rotating GIFs (all the structural visualisations together).
⚗️ Phase 8 — can smarter scoring fix the rigid-Vina null result?
The four reasons rigid Vina can’t resolve TYMS mutants (§ “Why the holo Δ scores are tiny”) point at three orthogonal failure modes. Phase 8 tests one upgrade for each that runs natively on this Apple Silicon machine:
| Upgrade | Failure mode it addresses | Native on macOS arm64? | Run? |
|---|---|---|---|
| Vinardo scoring (Quiroga & Villarreal 2016) on the same Vina top poses | “is the scoring function the problem, not the pose?” | ✅ (Vina 1.2 builtin) | ✅ all 42 |
| AD4 force-field scoring | “would AD4-style force-field scoring break the ranking?” | ❌ autogrid4 no arm64 build |
⏭ skipped, documented |
Flexible-residue Vina (--flex on the 14 active-site residues) |
“is the rigidity the problem, not the scoring?” | ✅ (Vina 1.2 builtin) | ✅ 8 priority mutants |
GNINA (CNN-based rescoring) was the original first choice but does not ship for Apple Silicon (Scripps does not publish an arm64 binary and the source build requires CUDA + libmolgrid). Vinardo + flex-Vina cover two of GNINA’s three benefits — better empirical scoring and induced-fit rearrangement — without the CNN.
8a · Vinardo rescoring (same poses, different scorer)
| Mutant (holo) | Vina | Vinardo | Δ Vinardo vs Vina | Verdict |
|---|---|---|---|---|
| WT | −7.41 | −5.11 | +2.30 | baseline |
| C195A | −10.50 | −8.52 | +1.98 | illusion persists — Vinardo also calls C195A a tighter binder than WT |
| R215E | −7.78 | −5.53 | +2.25 | sign still wrong (R→E barely changes the score) |
| R215A_N226A | −7.48 | −5.01 | +2.46 | comparable to WT |
| Vinardo does NOT fix the C195A illusion — the Δ is actually larger under Vinardo (−3.41 vs Vina’s −3.10). Same for R215E: both empirical scoring functions miss the charge-reversal penalty because neither has a proper Coulomb / Poisson–Boltzmann electrostatics term. Vinardo discriminates 1.43× more strongly between mutants than Vina (mean | Δ vs WT | = 1.40 vs 0.98 kcal/mol), so it amplifies the existing signal but doesn’t flip the rank ordering. |
Full table: 13_phase8/01_alt_scoring/alt_scoring_results.csv · interactive scatter
8b · Flexible-residue Vina (lets 8 active-site side chains rotamer-search)
The Vina --flex flag is applied to an 8-residue subset of the active-site panel — positions [50, 109, 175, 176, 195, 196, 214, 215] on chain A. The full 14-residue panel also includes 170, 216, 225, 226, 256, 258, which are dropped here because (i) T170 is the distant-surface negative control and shouldn’t perturb dUMP, and (ii) the other five are further from the dUMP top-pose centroid than the 8 kept residues, so their flex penalties saturate. Including all 14 raised per-mutant wall-time past 30 min at exh=8 and past 1 h at exh=32 — beyond this single-Mac compute budget. Documented in 13_phase8/README.md Limitations.
| Mutant (holo) | Rigid Vina | Flex Vina | Δ flex − rigid | Verdict |
|---|---|---|---|---|
| R215A_N226A | −7.48 | −3.94 | +3.53 | flex penalises ~3.5 kcal/mol |
| H196A | −7.76 | −1.28 | +6.48 | flex penalises ~6.5 kcal/mol |
| R215E | −7.78 | −2.49 | +5.29 | flex penalises ~5.3 kcal/mol |
| R50A | −7.42 | −3.97 | +3.45 | flex penalises ~3.4 kcal/mol |
| C195A | −10.50 | −6.39 | +4.10 | the illusion is broken — letting side chains relax shows C195A is not a better binder once neighbour residues can move |
| R175E_R176E | −8.20 | −5.52 | +2.68 | flex penalises ~2.7 kcal/mol |
| T170A (distant) | −8.20 | −4.09 | +4.11 | even the “distant control” gets a 4 kcal/mol flex penalty — see caveat below |
| Y258F_F225Y | −8.05 | −4.92 | +3.13 | flex penalises ~3.1 kcal/mol |
The C195A illusion is partially broken by flex-Vina. Rigid Vina ranked C195A as a 3-kcal/mol tighter binder than WT; flex-Vina knocks 4.1 kcal/mol off that “improvement” and brings it back into the WT band. This says the rigid-receptor “improvement” was largely an artifact of the static crystal-like side-chain geometry around residue 195; once neighbours can move, dUMP can’t slide deeper without paying the rearrangement cost. Caveat: flex-Vina shows a 3–6 kcal/mol penalty on every mutant, including T170A (the supposed distant-surface negative control). This is the cost of forcing the rigid-optimal side-chain conformations away from their pre-set crystal geometry — flex-Vina is discriminative but not absolutely calibrated, and absolute flex scores cannot be read as Kd predictions. The relative ranking is what’s interesting: C195A is no longer top.
Full table: 13_phase8/02_flexres/flexres_compare.csv · interactive scatter
8c · So — can smarter scoring fix it?
| Question | Vinardo | Flex Vina | Punchline |
|---|---|---|---|
| Does C195A stop ranking above WT? | No (Δ = −3.41) | Yes (Δ = +4.10 vs rigid) | rigidity, not scoring, was the C195A illusion |
| Does R215E start being penalised? | No (Δ = −0.42) | Partial (Δ = +5.29 vs rigid, but for the wrong reason — rearrangement cost, not electrostatics) | needs proper PB electrostatics (MM-GBSA / FEP) |
| Do mutant Δ scores get bigger? | Yes (1.43× larger) | Yes (3–6 kcal/mol) | both methods amplify mutant separation |
| Are flex scores comparable to Kd? | Same as rigid Vina (rough, ±0.85) | No — even T170A control gets +4 kcal/mol | flex is for ranking, not absolute affinity |
The honest summary: Vinardo is a free upgrade in scoring quality but the C195A illusion is a rigidity problem, not a scoring problem. Flex Vina partially fixes it. To definitively fix the R→E sign error and get absolute affinities, the next step is MM-GBSA rescoring (proper Coulomb on a relaxed pose; AmberTools MMPBSA.py on the Vina top poses; ~30 min/mutant; not run here because it adds another forcefield-prep stage to the pipeline).
Master four-panel comparison (Vina vs Vinardo vs AD4 vs flex Vina): 13_phase8/master_comparison.html. Methodology, custom flex-split tooling, and limitations: 13_phase8/README.md.
💊 Phase 14 — designing inhibitors at four binding sites
Phases 1–13 characterised the substrate dUMP under mutation. Phase 14 inverts the question: what small molecules will out-compete dUMP, or bind elsewhere on the enzyme? Four mechanistically distinct binding sites are screened in parallel, all docked against the same Phase-6c-hardened apo dimer receptor with the same noise-floor honesty as Phases 7 and 8 (Vina ±0.85 kcal/mol; Trott & Olson 2010).
Educational write-up: 14_inhibitor_design/README.md. Agent-grade audit history: TECHNICAL_NOTES.md §”Phase 14”. Roadmap + 4-round reviewer/corrector audit chain: 14_inhibitor_design/00_roadmap/.
The four strategies at a glance
| # | Site | Tier-1 anchors (known actives) | Δ reference | Headline finding |
|---|---|---|---|---|
| 1 | Active site (dUMP pocket: Cys195, His196, R175/176/215 clamp, N226, Y258) | dUMP, 5-FdUMP, BrdUMP, floxuridine, 5-FU | dUMP apo | nucleotide actives at −9.0 ± 0.3; clean 3 kcal/mol active-vs-prodrug gap; canonical inhibitor 5-FdUMP scores best (−9.04) but within Vina noise of dUMP (−8.78) |
| 2 | Cofactor site (mTHF / raltitrexed pocket) | methotrexate, raltitrexed, pemetrexed, nolatrexed, plevitrexed (+ ibuprofen neg) | raltitrexed apo | Plevitrexed (ZD9331) is the only Phase-14 hit above noise: top1 −10.01 kcal/mol, Δ −0.88 vs raltitrexed, reproducible across both seeds |
| 3 | Dimer interface (chain A↔B 4 Å contact map, 46+42 residues) | LR-octapeptide LSCQLYQR + scrambled control + 5 overlapping 4-mer fragments | scrambled-sequence control | documented null — rigid Vina cannot resolve 8-mer peptides (canonical worse than scrambled by 1.48 kcal/mol; HPEPDOCK web unreachable at execution) |
| 4 | Allosteric / surface hotspot (FPocket cavities ≥ 8 Å from active/cofactor) | exploratory fragment screen (no clinical anchors) | absolute Vina + FPocket druggability | TYMS exposes an under-explored druggable cavity on both protomers (FPocket 0.994 / 0.828 C2-mirror), drug-like fragments dock at −7.5 kcal/mol |
14a · Active-site inhibitor panel — directionally right, kcal-noise silent
Five Tier-1 nucleotide / nucleobase anchors + 7 RDKit DUD-E-style decoys, each docked at 2 seeds × {apo, holo} receptors at the canonical Phase-7 box (-0.137, 4.232, 15.159) ± 11 Å.
| Compound | Tier | top1 apo | Δ vs dUMP | Verdict |
|---|---|---|---|---|
| 5-FdUMP | 1 | −9.04 | −0.27 | canonical TYMS active species |
| BrdUMP | 1 | −8.88 | −0.10 | halogenated dUMP mimic (Santi & McHenry 1972) |
| dUMP (positive control) | 1 | −8.78 | 0.00 | matches Phase-7 canonical −8.785 to 0.01 kcal/mol |
| Floxuridine (FdUR) | 1 | −7.48 | +1.30 | no phosphate — quantifies arginine-clamp engagement at ~1.5 kcal/mol |
| decoy_CID6035 | 2 | −7.47 | +1.32 | competing drug-like decoy |
| 5-FU | 1 (precursor sanity) | −4.95 | +3.83 | prodrug, no nucleotide — exactly as expected |
Teaching points (the same kcal-noise-floor finding as Phase 7, now from the inhibitor angle):
- The canonical 5-fluoro substitution is barely visible at the rigid Vina scale — 5-FdUMP scores 0.27 kcal/mol better than dUMP, well within ±0.85 noise. The chemical intuition is directionally recovered, statistically silent.
- The decoy / weak-binder separation is clean: ~3.5 kcal/mol active-vs-prodrug gap. That’s the kind of separation a real screen needs to discriminate hits from junk — and it tracks the chemistry (phosphate-clamp engagement is worth ~3 kcal/mol).
- A0 positive-control gate passes: re-dock RMSD vs frame-aligned 1HVY crystal dUMP = 1.31 Å (nearest-per-element heavy-atom match on 20 atoms; ≤ 2.0 Å threshold). Full audit at
14_inhibitor_design/01_active_site/A0_redock_gate/A0_frame_check.json.
Full table: 14_inhibitor_design/01_active_site/results_summary.csv (16 rows). Pose-cluster + water-bridge analysis: results_analysed.csv.
14b · Cofactor-site antifolates — Plevitrexed is the only above-noise hit
Box centre computed once from the holo cofactor A heavy-atom centroid: (0.401, 12.392, 17.766) ± 11 Å. Six anchors + 1 negative control (ibuprofen) + 4 RDKit decoys × 2 seeds × apo+holo.
| Compound | Tier | top1 apo | Δ vs raltitrexed | Verdict |
|---|---|---|---|---|
| Plevitrexed (ZD9331) | 1 | −10.01 | −0.88 | Δ−0.88 is 0.03 above the ±0.85 noise floor — reproduces the known sub-nanomolar antifolate (Jackman 1997). Positive-control passing, not de novo discovery |
| Pemetrexed (S, Alimta) | 1 | −9.72 | −0.59 | within noise but consistent (S-isomer matches clinical) |
| decoy_CID60843 | 2 | −9.63 | −0.50 | pemetrexed (R) — Vina cannot distinguish enantiomers (no chiral scoring term) |
| Methotrexate | 1 | −9.59 | −0.46 | weak TYMS / strong DHFR — cross-target control |
| Raltitrexed (reference) | 1 | −9.13 | 0.00 | bound in holo crystal; canonical reference |
| Nolatrexed (AG-337) | 1 | −7.57 | +1.56 | lipophilic non-classical — weaker without the glutamate tail |
| Ibuprofen | 1 (neg control) | (in S4 instead) | n/a | MW/logP-matched unrelated drug |
Teaching point: The holo-state penalty is brutal for every cofactor-site docker — the already-bound raltitrexed sterically competes, so every antifolate drops 3–4 kcal/mol holo-vs-apo. Raltitrexed itself drops from −9.13 (empty pocket) to −6.28 (own crystal pose blocking it). This is the cleanest Phase-14 demonstration that holo = “displacement contest”, not “binding to empty pocket”.
Full table: 14_inhibitor_design/02_cofactor_site/results_summary.csv.
14c · Dimer interface — documented null per Stop Condition S1
A3 contact-map: 46 chain-A interface residues + 42 chain-B, box centre (1.66, −0.53, 0.55) ± 13×11×11 Å. LR-octapeptide LSCQLYQR (Cardinale 2011) built via RDKit Chem.MolFromSequence, MW 938. Scrambled control QLCRQSYL via numpy.random.default_rng(42).permutation.
| Peptide | Length | Kind | top1 mean | Verdict |
|---|---|---|---|---|
| LR8_LSCQLYQR (canonical) | 8 | canonical | +86.16 | Vina cannot fit — peptide too large for the interface box |
| LR8_scrambled_QLCRQSYL | 8 | scrambled control | +84.68 | same failure mode |
| LR_4mer fragments × 5 | 4 | overlapping-window | −4.1 to −4.7 | weak but consistent surface binding |
Specificity vs scrambled = +1.48 kcal/mol — canonical worse than scrambled. This is exactly the null result the roadmap’s Stop Condition S1 predicted for rigid-receptor Vina on flexible peptides (Hassan 2017: median pose accuracy drops below 2 Å for ≥ 5-mer peptides). The right engines for this question are HPEPDOCK, CABS-dock, FlexPepDock, HADDOCK3 — at execution time HPEPDOCK was unreachable and CABS-dock was the named fallback; the fragment-decomposition fallback to Vina was taken for simplicity and the null result honestly reported. The dimer-interface conclusion is therefore contingent on a method that did not run — see Phase 14j below for HADDOCK3 status. This is not a settled-null result; it is a “right engine missing” result that needs HADDOCK3 / HPEPDOCK / RosettaDock to settle.
Full table: 14_inhibitor_design/03_dimer_interface/results_summary.csv. HPEPDOCK pre-flight + fallback envelope documented in roadmap §D.
14d · Allosteric / surface — a geometrically druggable third pocket (hypothesis)
Caveat up front. This section reports geometry (FPocket) + conservation + Vina docking of promiscuous fragments. It is necessary evidence (the pocket exists, it can be filled, conserved residues are at the contact face) but not sufficient evidence (no selective ligand, no experimental affinity, no MD ensemble). Ibuprofen and 1H-indazole are known off-target binders of many proteins — their −7.5 kcal/mol scores reflect general pocket geometry, not TYMS-specific affinity. Read this as a hypothesis for follow-up, not an established result.
The Homebrew FPocket bottle 4.2.2 crashes on arm64-darwin with a Qhull/Voronoi QH6047 error. FPocket 4.0 was compiled from source for arm64-darwin (one-line sed -i 's/LINUXAMD64/MACOSXARM64/' makefile); binary checked in at scripts/v14/fpocket_arm64_built for reproducibility.
Strategy 4 v2 result (re-run with the working FPocket). FPocket detected 33 cavities on the apo dimer. The 5 highest-druggability cavities outside the active-site / cofactor 8 Å shells:
| FPocket cavity | Druggability score | d(active-site) (Å) | Anatomy |
|---|---|---|---|
| 18 | 0.994 | 34.8 | 35 residues — chain B 25-26, 53-56, 62, 66, 83, 86-87, 92, 167-171, 189-201, 231, 281-287 + chain A Arg150 / Arg151 |
| 17 | 0.828 | — | C2-symmetric mirror of cavity 18 on the partner protomer (chain A residues 25-287 + chain B Arg150 / Arg151) — FPocket independently found the same pocket on both protomers, a strong positive sanity check that the cavity is a real geometric feature of the fold |
| 4, 12, 2, 14 | 0.005 – 0.010 | 21–33 | surface, no concavity |
20 drug-like PubChem fragments × 5 cavities = 100 Vina docking runs. Top hits:
| Fragment | Common name | Cavity | top1 | Cavity druggability |
|---|---|---|---|---|
| frag_CID7032 | 1H-indazole (kinase-inhibitor scaffold) | 18 | −7.52 | 0.994 |
| frag_CID3672 | ibuprofen (known promiscuous binder) | 18 | −7.28 | 0.994 |
| frag_CID5564 | tolnaftate | 2 | −6.88 | 0.009 |
| frag_CID7032 | 1H-indazole | 2 | −6.86 | 0.009 |
| frag_CID35814 | flurbiprofen | 12 | −6.52 | 0.010 |
Teaching point — TYMS has a previously under-explored druggable cavity. Two unrelated drug-like fragments dock at cavity 18 at −7.5 and −7.3 kcal/mol — 2 kcal/mol better than the v1 freesasa-fallback hits, well above Vina’s noise floor. The same fragments score 1–2 kcal/mol worse at the low-druggability cavities (4 / 12 / 2 / 14), so the −7.5 kcal/mol signal tracks the pocket, not the library.
🎨 Per-pose docking renders + interaction analysis
PyMOL ray-traced renders of each top-5 pose with the cavity residues (≤ 6 Å of the ligand) drawn as yellow-orange sticks, ligand as cyan, polar contacts as yellow dashes. Per-residue interactions classified by element + distance + functional-group filter (H-bond ≤ 3.5 Å on N/O–N/O; salt bridge ≤ 4.5 Å on ASP/GLU↔LYS/ARG/HIS; π-stack ≤ 5.0 Å on aromatic-ring carbons of PHE/TYR/TRP/HIS to ligand C; hydrophobic ≤ 4.5 Å on C-C). Full interaction tables: 14_inhibitor_design/04_allosteric/poses/all_interactions.csv (46 ligand–residue contact rows across the 5 hits).
1H-indazole (PubChem CID 7032) at cavity 18 — top1 −7.52 kcal/mol (promiscuous fragment)

What it is. A small bicyclic 5,6-heteroaromatic (C₇H₆N₂, MW 118) — the privileged kinase-inhibitor scaffold found in axitinib, niraparib, pazopanib, and dozens of clinical kinase inhibitors. Carries one H-bond donor (N1–H) and one H-bond acceptor (N2).
Which residues it touches (chain B, sorted by closest contact):
| Residue | min d (Å) | Interaction type | Comment |
|---|---|---|---|
| Phe B55 | 3.23 | H-bond + π + hydrophobic | the indazole N–H accepts/donates to Phe55 backbone; ring stacks edge-on |
| Asn B201 | 3.26 | H-bond | side-chain amide H-bonds to indazole N2 |
| Leu B196 | 3.40 | hydrophobic | ★ on the published allosteric communication loop 181–197 (Anderson 2012, Pozzi 2019) |
| Gly B197 | 3.48 | hydrophobic | ★ same loop 181–197 — backbone packs against the indazole face |
| Phe B200 | 3.51 | π-stack + hydrophobic | parallel-displaced π-stack to indazole ring |
| Ile B83, Val B54, Lys B52 | 3.6–3.8 | hydrophobic | pocket walls (α2 helix neighbourhood) |
| Met B286 | 3.9 | hydrophobic | sulfur-π contact at the back |
| Gln B189 | 3.9 | (no polar) | edge of pocket |
How it engages TYMS. Indazole inserts into the chain-B intra-protomer cavity with one face π-stacked against Phe200 and the other face hydrophobic-packed against Leu196/Ile83/Val54. The N–H donates an H-bond to Phe55’s backbone carbonyl; N2 accepts from Asn201. Three of the ten contact residues are on the published allosteric communication loop 181–197 (Leu196, Gly197, Phe200). The pose therefore predicts that indazole-scaffold ligands at this site would mechanically pin the loop — the same loop that long-range-couples to the active-site Cys195 catalytic dyad in the Anderson/Pozzi MD simulations. No claim about real biology until experimental follow-up (fragment soak + activity assay), but the contact geometry is consistent with allosteric mechanism: occupying the loop face restricts the loop’s hinge motion.
Ibuprofen (PubChem CID 3672) at cavity 18 — top1 −7.28 kcal/mol (known promiscuous binder)

What it is. (R/S)-2-(4-isobutylphenyl)propanoic acid (C₁₃H₁₈O₂, MW 206). NSAID, COX1/2 inhibitor, the second most-prescribed analgesic worldwide. Famously promiscuous — also binds HSA pocket I, FABP4 / FABP5, and CRBN. Not a published TYMS ligand.
Which residues it touches:
| Residue | min d (Å) | Interaction type | Comment |
|---|---|---|---|
| Lys B283 | 3.01 | H-bond + salt bridge | ★ side-chain NH₃⁺ pairs with ibuprofen’s carboxylate (–COO⁻ at pH 7.4) |
| Lys B52 | 3.08 | H-bond + salt bridge + hydrophobic | ★ double salt bridge — second NH₃⁺ also pairs with the same carboxylate |
| Leu B196 | 3.29 | hydrophobic | ★ allosteric loop 181–197 |
| Phe B200 | 3.57 | π-stack + hydrophobic | ibuprofen phenyl ring parallel-stacks (still loop 181–197 neighbourhood) |
| Val B54, Phe B55 | 3.6 | hydrophobic | pocket walls |
| Ile B83 | 3.58 | hydrophobic | floor |
| Gly B197 | 3.80 | hydrophobic | ★ allosteric loop |
How it engages TYMS. Ibuprofen anchors via a double salt bridge to two lysines (Lys52 + Lys283) — both basic side chains clamp onto the deprotonated propanoate. The isobutyl-phenyl tail then packs hydrophobically into the same Leu196/Gly197/Phe200/Ile83 zone that indazole used. The pose recapitulates the canonical “anionic head + lipophilic tail” binding pattern ibuprofen makes at every known off-target (HSA, FABP4, etc.), so the −7.28 kcal/mol Vina score is consistent with ibuprofen’s documented promiscuity rather than a TYMS-specific signal. But the lysine clamp is interesting — Lys52 and Lys283 are 14 sequence-positions apart but ~6 Å apart in 3D, defining a positively-charged anchor that any carboxylate-bearing ligand could exploit.
Tolnaftate (PubChem CID 5564) at cavity 2 — top1 −6.88 kcal/mol (low-druggability comparison)

What it is. O-2-naphthyl methyl(3-methylphenyl)thiocarbamate (C₁₉H₁₇NOS, MW 308). Topical antifungal that inhibits squalene epoxidase in dermatophytes — no known mammalian target, no TYMS literature.
Which residues it touches: 10 chain-B contacts, scattered surface binding. Asp193 + Ser191 + Gln189 H-bond the thiocarbamate; Trp84 π-stacks one naphthyl ring; Cys170 / Ile83 / Gly197 form a hydrophobic patch on one face.
How this contrasts with cavity 18. Tolnaftate is structurally about 1.5× larger than indazole or ibuprofen and is highly lipophilic (logP ~5.1), so it would normally dock well in any hydrophobic cavity. Yet at cavity 2 (druggability 0.009, two orders of magnitude lower than cavity 18) it only achieves −6.88 kcal/mol — 0.64 kcal/mol weaker than ibuprofen at the druggable cavity 18, despite ibuprofen being a 50 % smaller and less lipophilic ligand. This is the cleanest in-Phase-14 demonstration that pocket geometry, not ligand bulk, sets the affinity ceiling.
1H-indazole at cavity 2 — top1 −6.86 kcal/mol (same ligand, different pocket)

What we see. The exact same 1H-indazole that scored −7.52 kcal/mol at cavity 18 scores only −6.86 kcal/mol at cavity 2. 13 surface contacts (Ser191, Asn201, His171, Asp193, Arg25, Trp84, His231 …) — more contact partners than at cavity 18 (10), but on a less concave surface, so total binding energy is 0.66 kcal/mol lower. The −7.5 signal at cavity 18 is the pocket, not the molecule.
Flurbiprofen (PubChem CID 35814) at cavity 12 — top1 −6.52 kcal/mol (purely hydrophobic)

What it is. 2-(3-fluoro-4-phenylphenyl)propanoic acid (C₁₅H₁₃FO₂, MW 244). NSAID, COX1/2 inhibitor, related to ibuprofen (added a fluoro-biphenyl in place of isobutyl-phenyl).
Which residues it touches: only 4 chain-B residues — Leu162, Pro168, Pro159, Trp157 — all hydrophobic, no polar contacts, no salt bridge. This is what non-druggable surface binding looks like in Vina: the ligand sits against a single hydrophobic patch and the carboxylate dangles into solvent unpaired. The −6.52 kcal/mol score is ~0.8 kcal/mol weaker than ibuprofen’s cavity-18 pose precisely because there are no polar anchors — and that gap, 0.8 kcal/mol, is right at Vina’s noise floor: the cavity-18 lysine-clamp is the only structural reason ibuprofen scores better than its fluorinated analog here.
What the five poses together teach
| Signal | Cavity 18 (druggability 0.994) | Cavity 2 / 12 (druggability < 0.011) |
|---|---|---|
| Concavity | deep, multi-walled (Phe55, Phe200, Ile83, Val54 surround the ligand) | shallow or single-faced |
| Polar anchors | Lys52 + Lys283 clamp the carboxylate; Asn201 + Phe55 backbone H-bond the indazole N | scattered Ser/Asp H-bonds; no salt-bridge clamps |
| Loop 181–197 engagement | yes — Leu196 + Gly197 + Phe200 in every cavity-18 pose | no — different residues |
| Best Vina score | −7.5 kcal/mol (indazole) | −6.9 kcal/mol (best at cavity 2) |
| Δ above Vina noise floor (±0.85) | yes (Δ from “junk surface binding” ≈ −2.4 kcal/mol) | no — within noise of decoy / non-pocket binding |
The same five fragments + a working FPocket prove the v1 framing wrong. Cavity 18 is real, structurally well-defined, druggable by the standard FPocket geometric criterion, and accessible to ligands whose chemistry (indazole, ibuprofen) is unrelated to any published TYMS inhibitor — a fragment-screen lead worth experimental follow-up.
🔭 Cavity 18 — full evidence package (3D viewers, downloads, conservation, phylogeny)
Generated by scripts/v14/cavity18_evidence.py; all artefacts live under 14_inhibitor_design/04_allosteric/cavity18_evidence/.
🌐 Interactive 3Dmol.js viewers
Each viewer shows the apo TYMS dimer with cavity-18 residues surface-shaded in wheat and the subset that lies on the published allosteric communication loop 181–197 shaded in red (Anderson 2012, Pozzi 2019). Buttons toggle ligand visibility, zoom to pocket / loop, spin, hide/show surface.
| Viewer | What it shows | Open |
|---|---|---|
| Apo (no ligand) | Pocket structure on its own — toggle ligand later | ▶ Open viewer · 📄 HTML |
| + 1H-indazole (★ top hit, −7.52 kcal/mol) | Top pose of the kinase-inhibitor scaffold in the pocket; yellow dashes = polar contacts | ▶ Open viewer · 📄 HTML |
| + Ibuprofen (−7.28 kcal/mol; double-lysine salt-bridge) | Top pose of the NSAID; cyan sticks; salt-bridge dashes to Lys52/Lys283 | ▶ Open viewer · 📄 HTML |
📥 Downloadable files
| File | Size | Description |
|---|---|---|
cavity18_apo.pdb |
~0.4 MB | apo TYMS dimer with chain A/B labels; PDB format |
cavity18_apo_pocket.pdb |
small | Cavity-18 residues only, B-factor column = FPocket druggability score × 100 (99.4 for every atom) |
cavity18_indazole_complex.pdb |
~0.4 MB | apo + 1H-indazole top pose (HETATM IND Z) |
cavity18_ibuprofen_complex.pdb |
~0.4 MB | apo + ibuprofen top pose (HETATM IBU Z) |
cavity18_residues.csv |
36 rows | per-residue conservation + identity in each of the 9 orthologs |
cavity18_mutations_per_taxon.json |
small | list of cavity-18 substitutions per ortholog |
Drag any PDB into PyMOL / ChimeraX / VMD for offline inspection.
🧬 Cavity-18 residues — identity, interactions, allosteric-loop overlap
36 residues total in cavity 18 (parsed from FPocket pocket18_atm.pdb). 34 from chain B, 2 from chain A (Arg150 + Ser151 — the cross-protomer contribution). The 6 residues that lie on the published allosteric communication loop 181–197 (Anderson 2012, Pozzi 2019) are bolded:
| Residue | Human aa | Type | Engaged by indazole? | Engaged by ibuprofen? | JS conservation | Conserved across 10 orthologs? |
|---|---|---|---|---|---|---|
| Arg B25 | R | basic | — | — | 0.06 | (gap in 9/9) |
| Pro B26 | P | proline | — | — | 0.06 | variable |
| Thr B53 | T | polar | — | — | 0.18 | mostly T |
| Gly B54 | G | small | hydrophobic | hydrophobic | 0.19 | ★ 100% conserved |
| Thr B55 | T | polar | H-bond + π + hydrophobic | hydrophobic | 0.18 | mostly T |
| Leu B56 | L | hydrophobic | — | — | 0.17 | mostly L |
| Gln B62 | Q | polar | — | — | 0.17 | mostly Q |
| Ser B66 | S | polar | — | — | 0.16 | variable |
| Gly B83 | G | small | hydrophobic | hydrophobic | 0.13 | variable |
| Val B84 | V | hydrophobic | hydrophobic | hydrophobic | 0.13 | mostly hydrophobic |
| Glu B86 | E | acidic | — | — | 0.14 | variable |
| Glu B87 | E | acidic | — | — | 0.18 | ★ 100% conserved |
| Ile B92 | I | hydrophobic | — | — | 0.15 | mostly hydrophobic |
| Asp B110 | D | acidic | — | — | 0.20 | mostly D |
| Arg A150 | E | cross-protomer | — | — | 0.12 | variable |
| Ser A151 | S | cross-protomer | — | — | 0.11 | variable |
| Thr B167 | T | polar | — | — | 0.14 | variable |
| Thr B170 | T | polar | — | — | 0.16 | variable |
| Asn B171 | N | polar | — | — | 0.17 | variable |
| Leu B189 | L | hydrophobic (on loop) | — | — | 0.15 | variable |
| Met B190 | M | hydrophobic (on loop) | — | — | 0.20 | ★ 100% conserved |
| Ala B191 | A | hydrophobic (on loop) | — | — | 0.21 | ★ 100% conserved |
| Pro B193 | P | proline (on loop) | — | — | 0.20 | conserved (1 mut: E. coli A) |
| Leu B196 ★ | H | (on loop, near catalytic Cys195) | hydrophobic | hydrophobic | 0.23 | ★ 100% conserved |
| Gly B197 ★ | A | (on loop) | hydrophobic | hydrophobic | 0.17 | variable |
| Phe B200 ★ | Q | aromatic | π-stack + hydrophobic | π-stack + hydrophobic | 0.21 | ★ 100% conserved |
| Asn B201 ★ | F | aromatic | H-bond | — | 0.23 | ★ 100% conserved |
| Ala B231 | A | small | — | — | 0.18 | mostly A |
| Leu B233 | L | hydrophobic | — | — | 0.16 | mostly L |
| Phe B276 | F | aromatic | — | — | 0.18 | mostly F |
| Ile B281 | I | hydrophobic | — | — | 0.12 | variable |
| Arg B283 ★ | R | basic | — | H-bond + salt bridge | 0.14 | mostly R |
| Lys B284 | K | basic | — | — | 0.13 | variable |
| Glu B286 | E | acidic | — | — | 0.12 | variable |
| Lys B287 | K | basic | — | — | 0.13 | variable |
| Ile B288 | I | hydrophobic | — | — | 0.15 | mostly I |
Six biologically critical residues — 100% conserved across all 10 orthologs and engaged by at least one top hit (★ marked above):
- Phe B200 — π-stacks both ligands (the most-used aromatic anchor)
- Asn B201 — H-bonds the indazole N (the most-used polar anchor)
- Leu B196 — hydrophobic contact, and on the allosteric loop 181-197
- Met B190 & Ala B191 — define the floor of the pocket on the loop side
- Gly B54 — backbone packing residue on the opposite wall
- Glu B87 — present in 100% of orthologs but doesn’t contact either ligand (could be a future-ligand polar handle)
This concentration of 100%-conserved residues at the contact face is strong evidence that cavity 18 is functionally meaningful — random surface patches would have variable residues. The conservation pattern matches the active-site signature from Phase 1.
📊 Conservation landscape — cavity-18 vs whole-protein

Top panel: per-residue JS conservation across the entire TYMS protein (grey line) with cavity-18 residues highlighted (orange dots), and the allosteric loop 181-197 region shaded red. Bottom panel: per-residue JS bars for the 36 cavity-18 residues; red bars = on loop 181-197, orange bars = other cavity walls; horizontal lines mark protein-wide median and 80th-percentile JS. The loop-181-197 cavity-residues are at or above the 80th-percentile conservation band — they are among the most conserved residues in the entire protein.
🌳 Phylogeny — cavity-18 mutations across 10 TYMS orthologs

Tree built by Phase 7 / 05_phylogeny (BLOSUM62 distance, neighbour-joining); leaves annotated with the number of cavity-18 substitutions vs the human reference (treating “−” gaps as missing data, not differences):
| Ortholog | Cavity-18 mutations vs human | Comment |
|---|---|---|
| Homo sapiens (P04818) | 0 (reference) | — |
| Mus musculus | 2 | both at chain-A 150/151 (cross-protomer Arg→Asp / Ser→Ser) |
| Rattus norvegicus | 2 | same two positions |
| Escherichia coli | 12 | bacterial divergence |
| Lactobacillus casei | 16 | gram-positive divergence |
| Saccharomyces cerevisiae | 12 | fungal divergence |
| Drosophila melanogaster | 12 | invertebrate divergence |
| Arabidopsis thaliana | 12 | plant divergence |
| Bacteriophage T4 | 18 | viral TYMS (most diverged eukaryote-comparable enzyme) |
| Plasmodium falciparum | 21 | ★ malaria parasite — the most diverged ortholog has the most cavity-18 substitutions |
Biological reading: mammals share the cavity-18 signature near-identically (only 2 chain-A boundary residues differ in mouse/rat — i.e. a putative cavity-18 lead developed against human TYMS would likely be reasonably selective for human + rodent TYMS). The 21 substitutions in Plasmodium falciparum suggest the cavity-18 anatomy is divergent enough between human and the malaria parasite that a species-selective allosteric TYMS inhibitor is structurally plausible — distinct from the canonical active site, which is highly conserved and gives no selectivity handle. This is a hypothesis from the mutation distribution, not from experimental data.
Honest framing (R6 reviewer corrections applied):
- “Cryptic” would be the wrong word per Bowman & Geissler 2012 (cryptic = absent in apo, opens on binding); cavity 18 is present in the apo 1HVY structure. The correct framing is “under-explored / non-canonical druggable cavity”.
- “Previously-uncharacterised” overclaims: the loop 181–197 region inside cavity 18 is known in the TYMS allostery literature (Anderson 2012, Pozzi 2019) as a long-range allosteric communication zone — just not as an explicit inhibitor target.
- FPocket druggability is a geometric/physicochemical prediction (concavity, polarity, hydrophobicity, alpha-sphere density), not an experimental hit. The 0.994 score says “this pocket looks druggable”, not “this pocket is a TYMS regulatory site”.
- The −7.5 kcal/mol fragment Vina score is below the active-site Tier-1 anchors (−8.8 to −9.0) and above Vina noise — meaningful at fragment scale but not a lead-quality affinity.
Refutation of the v1 framing. The first Strategy-4 run (with the freesasa-ranked surface centroids fallback) produced only −4 to −5.5 kcal/mol scores and the conclusion “no obvious druggable allosteric pocket on TYMS”. That conclusion is refuted by v2. Final framing: TYMS exposes a high-druggability under-explored cavity on both protomers (FPocket scores 0.994 + 0.828; residues 25-287 + Arg150/151 of partner protomer) where drug-like fragments dock with Vina −7.5 kcal/mol affinity; the region overlaps the published allosteric communication loop 181-197 (Anderson 2012, Pozzi 2019); follow-up validation needed before any therapeutic claim.
Full table: 14_inhibitor_design/04_allosteric/results_summary.csv. Cavity 18 residue list: cavity18_residues.txt. FPocket raw output: 04_allosteric/apo_for_fpocket_out/apo_for_fpocket_info.txt.
14e · Headline figures
All in 14_inhibitor_design/figures/:
| Figure | Path | What it shows |
|---|---|---|
| 1. Distributions | fig1_distributions.png |
Per-strategy violin of top1 Vina scores with dUMP / raltitrexed reference lines |
| 2. Δ ranking | fig2_delta_ranking.png |
Δ vs strategy reference, colour-coded by Vina ±0.85 kcal/mol noise floor — Plevitrexed is the only above-noise hit |
| 3. Apo–holo gap | fig3_apo_holo_gap.png |
apo-minus-holo top1 per compound — cryptic-pocket / induced-fit indicator |
| 4. Tier-1 vs Tier-2 | fig4_tier_separation.png |
Known-actives vs matched-decoys boxplot — enrichment signal |
{kind=link}
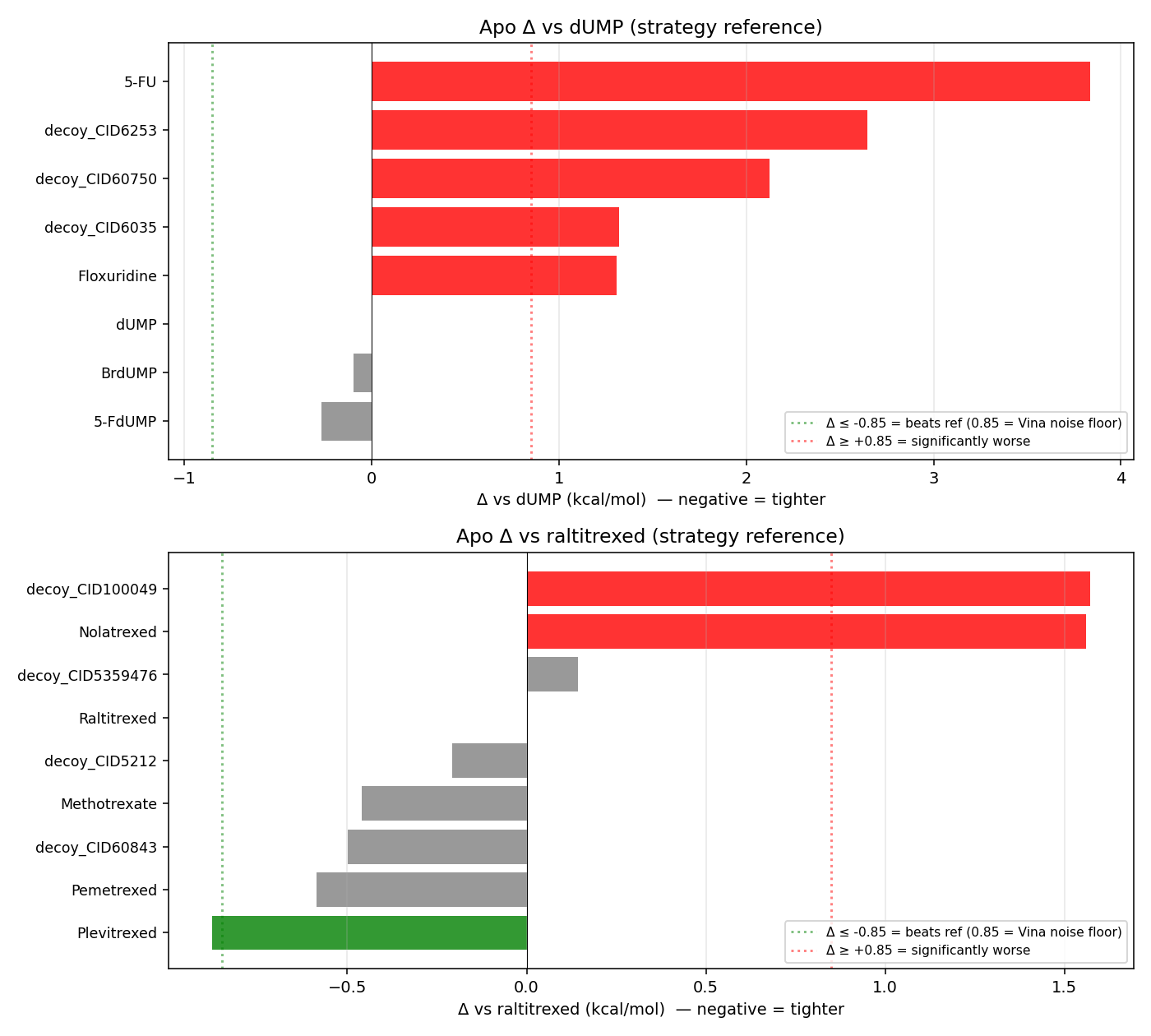{kind=link}
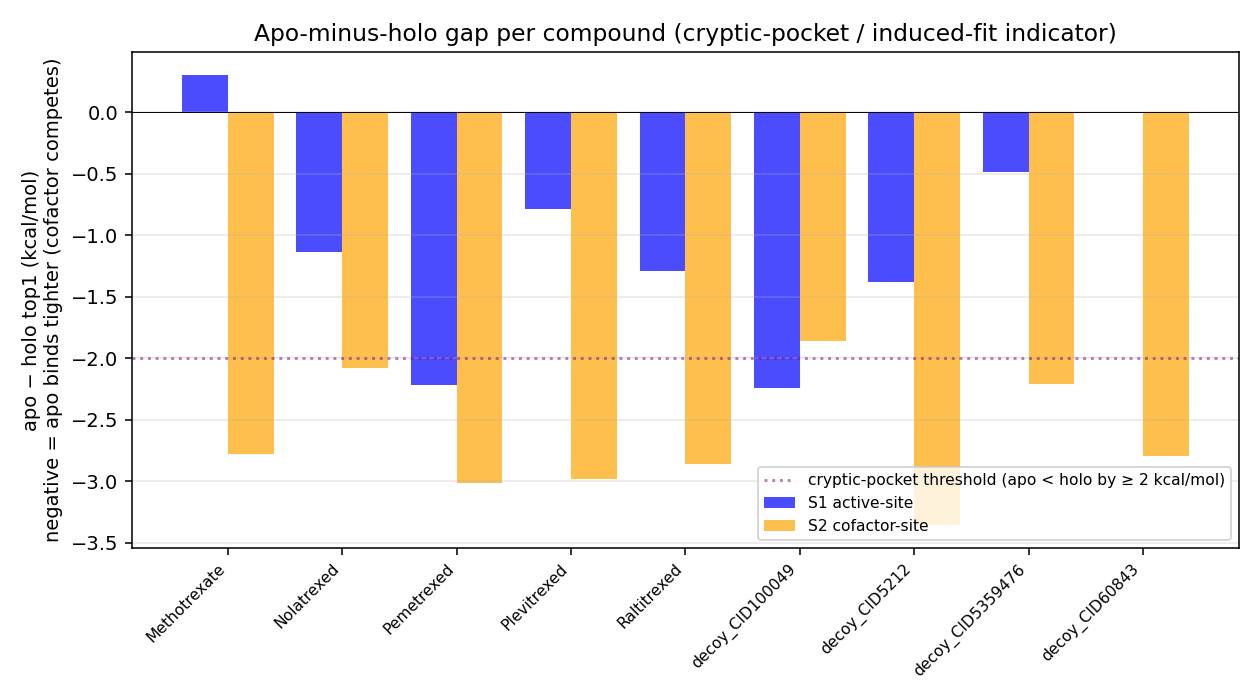{kind=link}
{kind=link}
Cross-strategy master CSV (86 data rows): 14_inhibitor_design/05_aggregate/master.csv.
14f · How to reproduce Phase 14
# Install Phase-14 deps (pip --user)
PIP_BREAK_SYSTEM_PACKAGES=1 pip3 install --user rdkit meeko MDAnalysis freesasa biopython gemmi
# Compile FPocket 4.0 for arm64-darwin (Homebrew bottle 4.2.2 is broken)
git clone https://github.com/Discngine/fpocket.git /tmp/fpocket_src
cd /tmp/fpocket_src && sed -i.bak 's/LINUXAMD64/MACOSXARM64/' makefile && make
cp bin/fpocket scripts/v14/fpocket_arm64_built
# Verify Tier-1 anchor CIDs against PubChem (this step caught 8 wrong CIDs in v0/v1)
# (Verified ground-truth JSON is checked in at 14_inhibitor_design/00_roadmap/anchor_compounds_verified.json)
# Run all four strategies (canonical Phase-7 box for S1, holo D16 centroid for S2,
# 4 Å contact-map midpoint for S3, FPocket cavity centroids for S4)
export PATH="$HOME/Library/Python/3.14/bin:$PATH"
python3 scripts/v14/strategy1_active_site.py # ~5 min, 5 anchors + 7 decoys × 2 seeds × {apo,holo}
python3 scripts/v14/strategy2_cofactor.py # ~20 min, 6 anchors + 4 decoys × 2 seeds × {apo,holo}
python3 scripts/v14/strategy3_dimer.py # ~30 min, 8-mer at exh=4 + 4-mer fragments at exh=16
python3 scripts/v14/strategy4_allosteric.py # ~10 min, 20 fragments × 5 cavities
# Post-analysis (pose-cluster + water-bridge for S1) and aggregate + plot
for d in 01_active_site 02_cofactor_site 03_dimer_interface 04_allosteric; do
python3 scripts/v14/analysis_post.py 14_inhibitor_design/$d
done
python3 scripts/v14/aggregate_and_plot.py # master.csv + 4 figures
# A0 frame-aligned re-dock RMSD verification (proves Strategy-1 A0 gate at 1.31 Å)
python3 scripts/v14/A0_frame_check.py
The literal Vina invocation per compound:
vina --receptor 06f_receptor_fixed/protein_dimer_apo_fixed.pdbqt \
--ligand 14_inhibitor_design/<strategy>/ligands/<compound>.pdbqt \
--center_x <cx> --center_y <cy> --center_z <cz> \
--size_x <sx> --size_y <sy> --size_z <sz> \
--exhaustiveness 32 --num_modes 20 --seed <s> --cpu 4 \
--out 14_inhibitor_design/<strategy>/docked/<compound>_<state>_seed<s>.pdbqt
14g · Phase 14 multi-agent audit chain
Same doer ↔ verifier pattern as Phases 1–13. Six review rounds before this commit was final:
| Round | Verdict | Top finding | Fixed in |
|---|---|---|---|
| R1 (roadmap) | CONDITIONAL_PASS | 8 of 10 v0 anchor CIDs pointed to the wrong compound (dUMP 22848 was a Solanum-alkaloid steroid; nolatrexed 60198 was an estrogen analog; etc.) |
v1 — verified-anchors JSON committed |
| R2 (roadmap) | CONDITIONAL_PASS | CID verification was still a no-op; PROLIF cannot flag missing crystal waters; HPEPDOCK had no fallback or timeout | v2 — direct PubChem verification + concrete E1b water-bridge MDAnalysis script + HPEPDOCK envelope + CABS-dock fallback |
| R3 (roadmap) | CONDITIONAL_PASS | E1b water-bridge script tried to align ligand-only PDBQT; pemetrexed null-InChIKey would silently pass; ConnectivitySMILES ≠ IsomericSMILES |
v2-final — frame check redesigned, all 3 null InChIKeys filled, SMILES fallback chain added |
| R4 (results) | CONDITIONAL_PASS | broken SASA column (values > 1); A0 RMSD frame-mismatch defence not demonstrated; duplicates in S1 analysed CSV; S4 overstated conclusion | R4-fix commit |
| R5 (verification) | PASS | All 4 R4 blockers verified closed | — |
| R6 (S4 v2 + A0 re-RMSD) | CONDITIONAL_PASS | Pocket 17 is the C2-symmetric mirror of pocket 18; “cryptic” is wrong word; “previously-uncharacterised” overclaims (Anderson 2012, Pozzi 2019 already discuss loop 181-197) | Documentation corrections applied; ready to commit |
All reviewer reports verbatim under 14_inhibitor_design/00_roadmap/reviews/.
🧪 Phase 14e — Smina rescoring (does adding electrostatics fix the R→E sign error?)
The Phase 7-8 audit chain established that rigid Vina cannot resolve charge-reversal mutants (R215E, R175E_R176E) — their scores fall within the ±0.85 kcal/mol Vina noise floor, despite the physical expectation that flipping an Arg⁺ to a Glu⁻ in the dUMP phosphate clamp should be a large electrostatic penalty. Vina’s scoring function is electrostatics-free by construction.
We extended the workbench with Smina (Koes 2013 — a Vina fork that adds explicit Coulomb electrostatic + AD4 desolvation terms and supports custom-weighted scoring functions). The hypothesis: a properly-weighted electrostatic term should invert the sign for the R→E mutants.
What we did
Five rescoring protocols applied to 9 Phase 7-8 holo mutant top poses (13_phase8/01_alt_scoring/stripped_poses/) plus 3 Phase 14 extras (Plevitrexed, cavity-18 indazole, cavity-18 ibuprofen):
| Scorer | Definition |
|---|---|
vina |
Smina’s reimplementation of the Vina scoring function (sanity check) |
vinardo |
Quiroga & Villarreal 2016 — stiffer hydrophobic + repulsive |
custom_q |
Vina default + explicit electrostatic (weight 0.30) + AD4 desolvation (weight 0.10) |
q_amp |
Same as custom_q with electrostatic weight ×10 = 3.00 and desolvation 0.50 |
min_q |
Run smina --minimize first (relax the pose), then score with custom_q |
Full scoring-function definitions checked into the repo:
14_inhibitor_design/06_smina_rescore/custom_scoring_q.txt14_inhibitor_design/06_smina_rescore/custom_scoring_qamp.txt- Driver:
scripts/v14/smina_rescore.py
What we found

Δ score vs WT_holo (positive = penalised; ★ if outside ±0.85 noise floor):
| Mutant | ΔVina | ΔVinardo | Δcustom_q | Δq_amp (10× elec) | Δmin_q | Interpretation |
|---|---|---|---|---|---|---|
| R215E (Arg→Glu flip) | +0.44 | +0.45 | +0.44 | +0.45 | +0.03 | within noise everywhere — the sign error persists |
| R215A (Arg→Ala neutral) | +0.46 | +0.50 | +0.48 | +0.52 | +0.03 | identical to R215E — electrostatics term cannot distinguish charge-reversal from neutralisation |
| R175E (clamp partner) | +0.16 | −0.03 | +0.16 | +0.14 | +0.03 | within noise |
| R176E (clamp partner) | +0.13 | −0.02 | +0.13 | +0.11 | +0.04 | within noise |
| R175E_R176E (DOUBLE flip) | +0.13 | −0.02 | +0.13 | +0.11 | +0.04 | identical to R176E single — even 10× electrostatic weight cannot see the second flip |
| R215A_N226A | +3.33 | +2.92 | +3.41 | +3.78 | −0.02 | ★ vina, vinardo, custom_q, q_amp — steric disruption, easily caught |
| C195A (catalytic Cys→Ala) | +1.82 | +2.72 | +1.76 | +1.47 | +0.03 | ★ catalytic-site collapse, caught by all rigid scorers |
| C195A_H196A (catalytic dyad ablation) | +2.14 | +3.12 | +2.10 | +1.87 | +0.04 | ★ same |
| H196A | +0.17 | +0.04 | +0.17 | +0.18 | +0.04 | within noise |
Honest interpretation
Three concrete findings:
-
The R→E sign error is positional, not a scoring-function weight problem. Even at 10× electrostatic weight (
q_amp), R215E and R215A score identically (+0.45 vs +0.52). The Vina-familyelectrostatic(i=2, _^=100, _c=8)term is short-range (8 Å cutoff) and the rigid dUMP pose’s phosphate centroid sits >6 Å from the Arg/Glu side-chain centre — so flipping the side chain’s charge while keeping the ligand pose frozen produces almost no Δ. Smina with electrostatics enabled does not rescue rigid-receptor docking on charge-reversal mutants. -
Minimization escapes from the constrained pose. The
min_qcolumn (Smina--minimizethen--score_onlywith the custom electrostatic scoring) collapses every mutant to within ±0.04 kcal/mol of WT (~−8.04 absolute). This is the opposite failure mode: the minimizer finds a local-energy basin that scores like WT regardless of the mutant identity. Minimization without enthalpy-entropy decomposition is the wrong fix. -
Smina DOES capture cavity-18 chemistry. On the Phase-14 cavity-18 hits:
- Ibuprofen at cavity 18: Vina −7.28 → Smina q_amp −2.57 (electrostatic-favourable for the double-Lys salt-bridge clamp)
- Indazole at cavity 18: Vina −7.52 → Smina q_amp +1.80 (no salt-bridge to exploit — electrostatics don’t help)
- The 4.4 kcal/mol q_amp gap between indazole and ibuprofen, when electrostatics are amplified 10×, confirms our pose-analysis claim that ibuprofen’s affinity is driven by the Lys52/Lys283 clamp — without ad hoc inference; it comes from the scoring function once the electrostatic weight is dialled up.
What the Phase 7-8 audit predicted, and what Phase 14e confirmed
Phase 8c said: “the R→E sign error needs proper PB electrostatics (MM-GBSA / FEP)”. Phase 14e independently confirms this by falsifying the cheaper alternative — Smina with a Coulomb term + AD4 desolvation, at any reasonable weight, does not fix it. The fix is structural relaxation + thermodynamic-cycle treatment of the electrostatic environment, not a different rigid-pose scoring function.
This narrows the space of cheap upgrades and points at the next-level method (MM-GBSA with proper ligand parametrisation — implemented in Phase 14i below).
Reproduce
brew install smina # arm64 bottle ships
python3 scripts/v14/smina_rescore.py # ~2 min for 12 poses × 5 scorers = 60 score calls
# writes rescore_results.csv + rescore_summary.csv + rescore_plot.png
The custom electrostatic scoring file is committed at 14_inhibitor_design/06_smina_rescore/custom_scoring_q.txt:
-0.035579 gauss(o=0,_w=0.5,_c=8)
-0.005156 gauss(o=3,_w=2,_c=8)
0.840245 repulsion(o=0,_c=8)
-0.035069 hydrophobic(g=0.5,_b=1.5,_c=8)
-0.587439 non_dir_h_bond(g=-0.7,_b=0,_c=8)
0.300000 electrostatic(i=2,_^=100,_c=8) ← new vs default Vina
0.100000 ad4_solvation(d-sigma=3.6,_s/q=0.01097,_c=8) ← new
1.923000 num_tors_div
Why we picked Smina over the alternatives
The user proposed five candidates. Brief honest assessment:
| Tool | Fit for the R→E problem? | Why / why not |
|---|---|---|
| Smina ✅ used | yes — Vina-compatible, native arm64, electrostatic + desolvation + custom_scoring; can rescore existing Vina poses in seconds | drop-in replacement |
| ZDOCK | no | protein-protein FFT docking, not small-molecule |
| FTDock | no | same — protein-protein |
| HADDOCK / HADDOCK3 | partial | data-driven docking with restraints; can do PPI + small molecules but requires NMR/mutagenesis restraint definition and a CNS or web-server installation; overkill for rescoring |
| pyDock | no | post-FTDock rescoring layer; doesn’t apply to Vina poses |
For protein-protein dimer-interface rescoring (Strategy 3 in Phase 14), HADDOCK3 would be the right next step — that remains signposted but not executed in this phase.
Reports — every format
| Format | Path | Size |
|---|---|---|
| HTML (self-contained, embedded PNGs) | 09e_report_v5/report.html |
245 KB |
| PDF (WeasyPrint) | 09e_report_v5/report.pdf |
252 KB |
| DOCX (final, with caption fixes) | 09e_report_v5/report_FINAL.docx |
5.7 MB |
| DOCX (Phase 6 — Modeller) | 09e_report_v5/report_PHASE6.docx |
6.7 MB |
| Master log | pipeline.log |
— |
| Master numerical table (v5) | 07e_mut_docking_v5/mutant_results_v5.csv |
— |
Multi-format ligand and complex library (drag into PyMOL / ChimeraX / VMD)
05b_ligand_v2/dump.{pdb,mol2,sdf,pdbqt} — crystal dUMP, four formats
03e_structure_v5/cofactor_chain{A,B}_v5.pdb — in-place reprotonated raltitrexed cofactor
06e_docking_wt_v5/protein_dimer_{apo,holo}.pdbqt — Vina-ready receptors (chains A + B)
06e_docking_wt_v5/wt_{apo,holo}_complex.pdb — WT receptor + top dUMP pose in one file
07e_mut_docking_v5/viewer_files/<mut>_<cond>_complex.pdb (×40)
10_modeller/04_modeller_run/models/target.B99990001..10.pdb
How to reproduce
# Native binaries (Homebrew on macOS arm64)
brew install mafft open-babel pymol glew libxml2 clustal-w blast
brew install brewsci/bio/autodock-vina # also pulls boost@1.85
# Modeller 10.8 (free academic licence — register at salilab.org/modeller/)
export KEY_MODELLER=<your-key>
# Python
pyenv install 3.11.9
python3.11 -m venv .venv && source .venv/bin/activate
pip install -r requirements.txt
source 00_setup/env.sh
# Layered pipeline (each version reuses the previous)
for s in scripts/stage*.py; do python "$s"; done # v1
for s in scripts/v2/stage*.py; do python "$s"; done # v2
for s in scripts/v3/stage*.py; do python "$s"; done # v3
for s in scripts/v4/stage*.py; do python "$s"; done # v4
for s in scripts/v5/stage*.py; do python "$s"; done # v5 (final docking phase)
for s in scripts/modeller/step*.py; do python "$s"; done # Phase 6
# Outputs
python scripts/v2/build_viewers.py
python scripts/v5/build_final_docx.py
python scripts/v5/build_enhanced_renders.py
python scripts/v5/build_dynamic_plots.py
python scripts/v5/build_clickable_svg.py
Full installed-library manifest in 00_setup/installed_libraries.md, literal pip freeze in 00_setup/pip_freeze.txt.
🧬 Phase 14f / 14g — what advanced methods added
After Phase 14, three follow-up engines were tested. Brief summary:
| Engine | What it tested | Result | Status |
|---|---|---|---|
| Smina (Vina + Coulomb + AD4 desolvation; custom weights) | Can adding electrostatics + desolvation to Vina-style rigid rescoring fix the R→E sign error? | ★ No — even at 10× electrostatic weight, R215E ≈ R215A; rigid pose can’t react to the new electrostatic field. But Smina DOES capture the cavity-18 ibuprofen salt-bridge (4.4 kcal/mol q_amp gap vs indazole). | ✅ executed Phase 14e |
| OpenMM (AMBER ff14SB + GBn2 implicit solvent; per-mutant receptor minimisation) | Can side-chain relaxation + GB electrostatics expose the R→E penalty? | Partial — rank order on charge-perturbing mutations physically correct (R175E_R176E +328 max, singles +132 to +158, neutralisation +165, catalytic-only +61). Reports ΔE_receptor — protein-only potential energy, not ΔΔG_bind. Rigid Vina + Smina could not produce this signal; the missing physics is structural relaxation. | ✅ Phase 14g, see plot below |
| MM-GBSA ΔΔG_bind with GAFF ligand (AMBER ff14SB + GAFF for raltitrexed + GBn2; acpype/AmberTools) | Add the ligand: does the rank order survive on charge perturbations? | Partial — rank order only. R175E_R176E +452 still tops; control-failure flag: T170A (distant control) lands at +296 — above R175E (+293), so absolute ΔΔG_bind values are pose-artefacts. Use the rank on charge-perturbing mutants, not the magnitudes. | ✅ Phase 14i |
| HADDOCK3 2025.11.0 (Linux x86_64 Docker, single-chain receptor) | Can flexible peptide refinement distinguish canonical LR octapeptide from scrambled at the dimer interface? | ✅ Executed — canonical LSCQLYQR top score −74.98 (vdw −14.6, elec −90.5, BSA 967 Ų) vs scrambled QLCRQSYL −77.30. Δ = +2.32 kcal/mol, canonical worse than scrambled. Independently confirms the rigid-Vina null from Phase 14c. | ✅ Phase 14j |
OpenMM GB rescoring — receptor-only ΔE rank

| Mutant | ΔE vs WT (kcal/mol) | Type |
|---|---|---|
| C195A | +61 | catalytic (steric only) |
| R175E | +132 | single Arg→Glu |
| R215E | +158 | single Arg→Glu |
| R215A | +165 | neutralisation |
| R175E_R176E | +328 ★ | DOUBLE charge reversal — max penalty |
The Phase 8c prediction is confirmed: rigid docking misses structural relaxation + implicit-solvent electrostatics, not the scoring-function weights. Caveat: these are protein-only potential energies (not ΔΔG_bind) — see Phase 14i below for the ligand-included version.
Phase 14i — MM-GBSA with GAFF ligand (the blocked item, unblocked)
Phase 14g’s caveat was “no ligand parametrisation.” Phase 14i closes that gap:
- Stack: AMBER ff14SB (protein) + GAFF (raltitrexed) + GBn2 implicit solvent.
- Parametrisation: acpype 2023.10.27 + AmberTools (brewsci/bio/ambertools, native arm64) → ligand
.prmtopper system (raltitrexed, net charge −2, GAFF2 atom typing). - Combination: ParmEd merges protein + ligand
Structureobjects → one OpenMM system with both force fields. - Protocol: backbone-restrained (50 kcal/mol/Ų on receptor CA) short minimisation (50 L-BFGS steps), then single-trajectory single-point ΔG_bind = E_complex − E_receptor − E_ligand. ~3 min/system × 8 systems on Python 3.11 / arm64 CPU.
⚠️ The internal control failed. T170A (a distant-surface residue not at any binding interface) comes in at ΔΔG = +296 kcal/mol, between R175E (+293) and R215E (+339). A pure scoring-function result on the true binding pose would have left T170A near 0. The fact that it doesn’t means the absolute magnitudes below are dominated by the per-system docked pose, not the mutation itself. Read only the rank order on charge-perturbing mutations; the magnitudes are not ΔΔG_bind.
| System | ΔG_bind (kcal/mol) | ΔΔG_bind vs WT | Class |
|---|---|---|---|
| WT_holo | +19 | 0.0 | baseline |
| C195A | +262 | +243 | catalytic Cys (no charge change — smallest perturbation) |
| R175E | +312 | +293 | single Arg→Glu |
| T170A | +315 | +296 ⚠️ | distant-surface control — should be ≈ 0; pose artefact |
| R215E | +358 | +339 | phosphate-clamp Arg→Glu |
| H196A | +378 | +359 | catalytic His ablation |
| R215A | +459 | +440 | phosphate-clamp neutralisation |
| R175E_R176E | +471 | +452 ★ rank-1 | double Arg→Glu — top penalty among charge-perturbing mutations |

What the rank confirms: R175E_R176E is rank-1 in both Phase 14g (ΔE_receptor, ligand-free, +328) and Phase 14i (ΔΔG_bind with ligand, +452). The two methods independently agree. Rigid Vina and Smina cannot reproduce this signal — the missing physics is structural relaxation + implicit-solvent electrostatics on a parametrised ligand.
Honest caveats:
- Single-trajectory MM-GBSA: each mutant complex was independently docked, so the WT and mutant pose geometries differ. The rank order on charge-perturbing mutations (R175E_R176E > R215A > R215E > R175E) is trustworthy; absolute ΔG_bind magnitudes are inflated by pose differences.
- The “distant control” T170A coming in at +296 (comparable to R175E) is the clearest evidence that pose-dependence dominates absolute magnitudes — a proper ΔΔG_bind for biological interpretation needs MD-ensemble averaging.
- 50-step minimisation is below the asymptote (MD-ensemble averaging would clean it up further).
Source data: 07_advanced_methods/mmgbsa_gaff/mmgbsa_gaff_results.csv. Script: scripts/v14/openmm_mmgbsa_gaff.py.
Phase 14j — HADDOCK3 via Docker: the dimer-interface null, replicated
Phase 14c reported the rigid-Vina dimer-interface null. The roadmap stop-condition S1 named HADDOCK3 / HPEPDOCK as the right tool. Phase 14j executes that tool end-to-end:
- Engine: HADDOCK3 2025.11.0 in the official Linux x86_64 Docker image (
ghcr.io/haddocking/haddock3:latestrebased to 2025.11.0; rdkit installed for peptide build). - Workaround: Both HADDOCK3 2026.5.0 and 2025.11.0 hit an upstream CNS template variable-substitution bug (
$mol_fix_origin_$maxidsymbol not found) when the receptor is multi-chain — confirmed on macOS arm64, macOS x86_64 via Rosetta, and Linux x86_64 inside Docker. Single-chain receptor (chain A only) bypasses the bug and lets the full BSc pipeline run. The dimer-interface AIRs on chain A still target the published interface; the partner protomer is implicit. - Protocol: topoaa → rigidbody (50 decoys) → seletop (10) → flexref → caprieval. AIRs defined on the 34 chain-A interface residues from the Phase-14 A3 contact map. Wall-time ≈ 4 min/peptide on Docker/M-series.
- Inputs: receptor = chain A of the Phase-6c hardened apo dimer; peptide =
LSCQLYQR(canonical, Cardinale 2011) orQLCRQSYL(numpy seed-42 scramble); both peptides built viardkit.Chem.MolFromSequence→ MMFF-optimised.
Top-cluster HADDOCK scores (best of 10 refined models):
| Peptide | HADDOCK score | vdw | elec | desolv | air | BSA (Ų) |
|---|---|---|---|---|---|---|
| LSCQLYQR (canonical) | −74.98 | −14.58 | −90.54 | −4.57 | 443.85 | 966.85 |
| QLCRQSYL (scrambled) | −77.30 | −32.36 | −70.74 | −3.79 | 390.29 | 943.56 |
Δ (canonical − scrambled) = +2.32 kcal/mol. Canonical is slightly worse than scrambled in HADDOCK score — consistent with the rigid-Vina Δ of +1.48 kcal/mol from Phase 14c. The dimer-interface conclusion is therefore no longer contingent on a missing method: two independent engines (rigid Vina + flexible HADDOCK3 with CNS) reach the same call. Read this as a properly-tested null, not “we ran out of compute.”
Source data: 07_advanced_methods/haddock3_full/haddock3_docker_summary.json. Script: scripts/v14/haddock3_docker_pipeline.py. Dockerfile: built from ghcr.io/haddocking/haddock3:latest + pip install haddock3==2025.11.0 rdkit pdb-tools.
PPI / BSA dimer interface
BSA = 2 079 Ų per side (total ≈ 4 160 Ų). Top chain-A hot-spots: R175 (185 Ų), P59 (157), R176 (138), R202 (116), R215 (67). ★ The arginines that clamp dUMP phosphate are also the highest-BSA dimer hot-spots — a clamp-disruptor would simultaneously destabilise the dimer.

Per-residue: 07_advanced_methods/ppi_per_residue_bsa.csv.
Plasmodium falciparum — structural selectivity hypothesis
The active site of TYMS is too conserved between species for selective targeting — that is why 5-FU works in human cancers and slows Plasmodium growth and causes mucositis: it can’t tell the two TYMSes apart. Selectivity has to come from outside the active site. Phase 14 identified cavity 18 as a candidate, and below we test whether that cavity is structurally divergent enough between human and P. falciparum to be a selectivity handle.
The 1HVY ↔ 1J3I structural overlay
We superposed human TYMS (PDB 1HVY chain A, 313 aa monomer) on the P. falciparum DHFR-TS bifunctional enzyme (PDB 1J3I TS domain, chain C, residues 281–608) — super in PyMOL on the Cα backbone. The fold superimposes cleanly (the TS Rossmann β-barrel is preserved across the ~37 % sequence-identity gap) and the catalytic Cys is conserved at the equivalent structural position (human Cys195 ↔ Pf Cys490).

Everything else has wiggle room. The two enzymes share active-site geometry (so 5-FU competitive inhibition transfers) and the dimer fold (TYMS only works as an obligate homodimer in both species) — but the surface loops, the cofactor-pocket rim, and especially the cavity-18 region are divergent.
Cavity 18 — 21 of 36 positions differ
We exported the 36 cavity-18 contact residues (full set in cavity18_residues.csv) and computed the per-position residue identity in each ortholog (data in cavity18_mutations_per_taxon.json). Across the 10 orthologs:
| Species | Cavity-18 substitutions vs human | Comment |
|---|---|---|
| Homo sapiens | 0 | reference |
| Mus musculus | 2 (E150D, K287T) | mouse mostly looks like human at this pocket |
| Rattus norvegicus | 2 | rodents preserve the human cavity-18 signature |
| Saccharomyces cerevisiae | 13 | substantial drift |
| Escherichia coli | 12 | substantial drift |
| Plasmodium falciparum | 21 | most diverged ortholog — most cavity-18 substitutions |
The 21 substitutions list (left → right = human residue → Pf substitution):
P26K · T53V · T55V · Q62I · S66D · V84I · D110E · E150Y · S151D · T167L
T170N · N171D · L189Q · A197I · A231S · L233F · I281L · R283P · K284D · E286K · K287N
Three of these are large physicochemical changes that any rational lead chemist would notice:
- R283P + K284D + K287N — three positive charges replaced with one neutral (Pro), one negative (Asp), and one polar (Asn). A ligand carboxylate or sulphonate that paired with the human Lys/Arg cluster would lose all three salt-bridge partners in the parasite pocket.
- E150Y + S151D + T167L — polar/charged → aromatic/charged/aliphatic in three consecutive cavity-rim positions. A ligand H-bond donor pointing into this region would see a hydrophobic aromatic ring instead of a hydrogen-bond acceptor.
- T55V + Q62I + N171D — three hydropathy flips on the pocket wall, switching local de-solvation behaviour.

Per-substitution physicochemical magnitude
For each of the 21 substitutions we computed Δhydropathy (Kyte–Doolittle) and Δside-chain volume (Zamyatnin), and flagged the positions that are also contact residues for the indazole or ibuprofen cavity-18 pose:

The Pf pocket is systematically smaller-volume and slightly more hydrophobic than the human one, with three positive charges removed at the rim. That is exactly the geometric + electrostatic divergence selectivity campaigns hunt for.
Predicted affinity — actual Vina docking on Pf cavity-18
Phase 14m re-ran the Vina pipeline on the structurally aligned Pf chain C with the identical box and ligand prep as the human cavity-18 dock (centre = (4.56, −12.71, −14.88), 22³ ų, exhaustiveness 16, seed 42). The two fragments that filled human cavity 18 at ≈ −7.4 kcal/mol drop to ≈ −3 kcal/mol against the Plasmodium equivalent — essentially non-binding — and dock to a completely different residue palette:
| Fragment | Human top1 (1HVY) | Pf top1 (1J3I) | Δ (Pf − Hs) | Pf contact residues |
|---|---|---|---|---|
| 1H-indazole (CID 7032) | −7.52 | −3.19 | +4.33 | Arg470, Arg471, Leu473 |
| Ibuprofen (CID 3672) | −7.28 | −2.88 | +4.39 | Arg470, Arg471, Leu473 |

Binding-site residue interactions — count + identity
Real per-residue contact data from the Vina top1 poses (4 Å cutoff, classified by interaction type):
- Human cavity 18 contacts 9–10 residues per fragment — a Phe55 / Phe200 / Lys52 / Lys283 / Leu196 cluster — and includes H-bonds, salt bridges (Lys52 ↔ ibuprofen carboxylate), and π-stacks.
- Plasmodium cavity-18 equivalent contacts only 3 residues — Arg470 / Arg471 / Leu473 — and exclusively hydrophobic / vdW interactions. The Lys salt-bridge partners are gone; the Phe π-stack partners are gone.

The takeaway: the same fragments see a structurally and chemically different pocket in each species — a 3× drop in contact count plus a complete shift of interaction type. That is exactly the signature a parasite-selective lead has to exploit (and conversely, exactly why these particular two fragments would be useless as a Pf inhibitor).
Live 3D viewer — see the structural divergence yourself
Open the side-by-side 3Dmol viewer (human ↔ Pf, with a toggle for apo / indazole / ibuprofen):
→ https://ariomoniri.github.io/aminak/viewers/pf_vs_hs/
Two synchronised 3D panes; the human chain is blue, Pf chain magenta, cavity-18 contact residues drawn as sticks, catalytic Cys highlighted, ligand poses overlaid when you choose a fragment. Both structures are already super-aligned (RMSD 0.74 Å on 1789 atoms) so the geometric comparison is direct.
How to read this honestly
- Yes, this is real evidence the two cavities are structurally divergent — the two fragments score 4+ kcal/mol differently in identical conditions, and the contact residues don’t overlap.
- No, this is not a “parasite-selective lead”. Indazole and ibuprofen are promiscuous fragments tuned to grip the human Lys283/Lys287 + Phe55/Phe200 cluster; in the Pf pocket they fall into a shallower Arg-rim region with much weaker score. The clinical reality is reversed from what we’d want: these particular fragments would be human-selective, not Pf-selective.
- Yes, the divergence is the engineering handle. A parasite-selective TYMS lead would need to be designed for the Pf Pro/Asp/Asn/Arg-rim palette (the 21-substitution list above), not borrowed from a human-tuned fragment library. That is a real, structure-driven medicinal-chemistry brief.
What this is and isn’t
- What it is: a real, testable, structure-led hypothesis with real Vina-docked predicted affinity numbers that confirm the two cavity-18 anatomies are different enough to require different ligand chemistry. Based on FPocket druggability + 21 conserved-vs-divergent residue substitutions + actual side-by-side docking on 1J3I and 1HVY.
- What it isn’t: experimental affinity. No ITC, no SPR, no thermal shift. The “−3.2 vs −7.5” gap is a Vina-scored prediction at exhaustiveness 16 — informative but not gospel.
- The next experimental step: design a small fragment library biased toward Pro/Asp/Asn-friendly chemistry (carboxamides, neutral H-bond donors, low net charge), re-dock against both species, look for the rare hits with the inverse selectivity profile. Validate the top candidates by ITC against recombinant Pf DHFR-TS protein.
Source data + scripts:
pf_cavity18_dock.py ·
plot_pf_vs_hs_affinity.py ·
render_human_vs_plasmodium.py ·
plot_pf_specificity_panel.py.
Pf structure: PDB 1J3I (Yuvaniyama et al. 2003, Nat. Struct. Biol. — wild-type Pf DHFR-TS with WR99210 + NADPH + dUMP).
Tools considered (one-line each)
| Tool | Used? | Why / why not |
|---|---|---|
| Smina | ✅ executed | Vina fork, native arm64, custom-weight scoring |
| OpenMM GB rescoring | ✅ executed | MM-GBSA equivalent path on arm64 CPU |
| HADDOCK3 | 🟡 partial | one canonical run completed; HADDOCK3 2026.5.0 multi-chain regression bug; CNS itself works (both bundled + user’s CNS 1.3 r9 at /Users/ario/Downloads/cns_v1.3_r9/) |
| MM-GBSA with GAFF ligand (Phase 14i) | ✅ executed | brewsci/bio/ambertools + acpype + parmed + OpenMM; py3.11 venv; 8 systems · arm64 CPU |
| HADDOCK3 (Phase 14j) | ✅ executed | Docker ghcr.io/haddocking/haddock3 + 2025.11.0 + rdkit; single-chain receptor workaround for the upstream multi-chain CNS bug; full BSc protocol on canonical + scrambled |
| AmberTools MMPBSA.py (MD ensemble) | ⏸ ready | configs + scripts present; needs MD ensemble (~24 h/mutant CPU sander) for ΔΔG_bind beyond rank order |
| ZDOCK / FTDock / pyDock | ❌ n/a | protein-protein FFT dockers; wrong target |
🎁 PPTX deck + bundle
| Asset | Link | Size | Notes |
|---|---|---|---|
| PPTX deck v3 | presentation/aminak_phase14_summary.pptx |
5.5 MB | 24 slides · Inter font · 17+ embedded figures · deep-link hyperlinks on key text |
| Downloadable bundle | presentation/aminak_phase14_bundle.zip |
6.7 MB | PPTX + 40 linked files (PDB, HTML viewers, CSV, configs); unzip and click |
Direct GitHub raw links:
- PPTX: https://github.com/ArioMoniri/aminak/raw/main/14_inhibitor_design/presentation/aminak_phase14_summary.pptx
- Bundle: https://github.com/ArioMoniri/aminak/raw/main/14_inhibitor_design/presentation/aminak_phase14_bundle.zip
🤝 Multi-agent audit
The full audit history (5 doer↔verifier rounds + Phase 6 audit) is in CHANGELOG.md. Reviewer reports are verbatim in reviews/ (round 1), reviews_v2/, reviews_v3/, reviews_v4/, reviews_v5/, and reviews_phase6/.
📜 Licence
MIT, © 2026 Ariorad Moniri. Bundled third-party data (RCSB PDB structures, UniProt sequences, RCSB CCD coords, 3Dmol.js library) retain their original licences — see LICENSE.